

IL CENTRO ASCOLTO NELLE MARCHE
 Grazie ad un finanziamento della Regione Marche è stata aperta una nuova sede del nostro centro ascolto a Gabicce Mare (PU).
Grazie ad un finanziamento della Regione Marche è stata aperta una nuova sede del nostro centro ascolto a Gabicce Mare (PU).
COMUNICATO 27.03.08
Apre nelle Marche la nuova sede del Centro Ascolto per la distrofia di Duchenne, un servizio per le famiglie e gli specialisti.
Apre a Gabicce Mare, in provincia di Pesaro e Urbino, la nuova sede del Centro Ascolto Duchenne grazie alla Regione Marche che, sulla base della LR 9/04, ha finanziato il progetto "Centri d'ascolto sulla Distrofia di Duchenne e Becker" dell'associazione di genitori Parent Project Onlus, che consentirà di potenziare le attività di ascolto già realizzate a livello nazionale.
"Non esiste attualmente un Network "coordinato e attivo esclusivamente" sulla distrofia muscolare di Duchenne e Becker (DMD/DMB). - ha dichiarato Sabrina Banzato, Delegato Regionale delle Marche - Gli enti locali della nostra Regione si stanno muovendo nella direzione giusta, presto sarà attivo il "Centro Regionale per le malattie neuromuscolari", organizzato dall'Azienda ospedaliera Ospedali riuniti di Ancona e con il quale speriamo di cominciare a collaborare da subito. Per le malattie rare, come nella Duchenne/Becker, la diffusione delle pratiche terapeutiche può consentire di raddoppiare l'aspettativa di vita dei ragazzi. In attesa che in tutto il territorio nazionale si elaborino strumenti capaci di rispondere ai bisogni delle famiglie che non trovano risposta a causa della carenza di conoscenze e competenze specifiche, sia da parte dei servizi sociali che sanitari, Parent Project continuerà diffondere il modello del Centro Ascolto Duchenne." - ha concluso il Delegato
Gli operatori del CAD, regionali e nazionale, agiranno in modo organizzato e strutturato per sostenere e sostenersi nella ricerca di risposte mirate e competenti al fine di migliorare la qualità dell'offerta. Questo sarà possibile grazie ad un apposito sistema informativo del lavoro sociale via web (denominato SILS), realizzato dalla SocialNet di Pesaro, che consentirà la gestione integrata, anche a distanza, delle situazioni su cui intervenire di comune intesa, migliorando così anche la qualità del lavoro.
Tra gli obiettivi previsti la costruzione di accordi di rete territoriale regionale (sui territori di tutte e quattro le province coinvolte dal progetto Pesaro e Urbino, Ancona, Macerata ed Ascoli Piceno) per la conoscenza e diffusione della malattia e l'accoglienza integrata delle famiglie.
Il CAD regionale, in particolare, fornirà informazione specialistica sulla malattia presso i servizi socio-sanitari della Regione Marche grazie anche ad incontri specifici e alla diffusione di materiale inviato ai servizi stessi e a disposizione delle famiglie residenti nelle quattro province.
La Distrofia Muscolare di Duchenne e Becker (DMD/DMB) è una  malattia rara, la forma più grave delle distrofie muscolari, che viene trasmessa dalle madri attraverso il cromosoma X e colpisce 1 su 3.500 maschi nati vivi. Attualmente non esiste una cura specifica, ma un trattamento da parte di una equipe multidisciplinare che ha permesso di migliorare le condizioni generali e raddoppiare le aspettative di vita. Si stima che in Italia ci siano 5.000 persone affette dalla patologia.
malattia rara, la forma più grave delle distrofie muscolari, che viene trasmessa dalle madri attraverso il cromosoma X e colpisce 1 su 3.500 maschi nati vivi. Attualmente non esiste una cura specifica, ma un trattamento da parte di una equipe multidisciplinare che ha permesso di migliorare le condizioni generali e raddoppiare le aspettative di vita. Si stima che in Italia ci siano 5.000 persone affette dalla patologia.
Parent Project Onlus, l'associazione fondata dai genitori con figli affetti dalla distrofia muscolare di Duchenne e Becker, nasce nel 1996 per sconfiggere questa grave distrofia muscolare promuovendo e finanziando la ricerca scientifica. E' socio fondatore della Federazione Internazionale UPPMD (United Parent Projects for Muscolar Dystrophies Ltd.). In questi anni ha contribuito, con oltre 1.500.000 euro di investimenti, al finanziamento di più di 50 progetti specifici. Altro obiettivo primario dell'associazione è la formazione e il sostegno per le famiglie coinvolte dalla malattia che viene erogato gratuitamente dal CAD che si può contattare al Numero Verde 800 943 333. Il servizio è attivo, dal 2002, grazie all'ISMA (Istituti Santa Maria in Aquiro di Roma) che contribuisce alle attività della sede nazionale e alla Fondazione Peppino Vismara che ha contribuito all'apertura della sede di Bergamo.
Il Centro Ascolto Duchenne si può contattare ogni giorno ed eventualmente prendere contatto con l'assistente sociale Ilaria Nanni presso la sede regionale in Via Romagna, 53 a Gabicce Mare (PU).
Per le iniziative realizzate nella Regione Marche contattare il Delegato Regionale Sabrina Banzato al numero 0541/410083. Per maggiori informazioni sulle attività di Parent Project visitate il sito internet www.parentproject.org
Ufficio stampa Parent Project Onlus
Stefania Collet
Tel. 06 66.18.28.11 - Fax 06 66.18.84.28
e-mail ufficiostampa@parentproject.org
COMUNICATO 27.03.08
Gli organizzatori del Rally di Alba impegnati insieme a Parent Project per sconfiggere la distrofia muscolare di Duchenne e Becker.
L'appuntamento con i volontari è il 5 e il 6 aprile, in piazza Medford ad Alba.
Prosegue l'impegno del mondo del rally piemontese al fianco di Parent Project, l'associazione di genitori che combatte contro la distrofia muscolare di Duchenne e Becker, che sarà ospite del "Rally di Alba" che si disputa dal 4 al 6 aprile 2008 nella magnifica cittadina piemontese. Il Cinzano Rally Team A.S.D., infatti, ha deciso di sostenere l'associazione per realizzare una raccolta fondi necessari a finanziare la ricerca di una cura per oltre 5000 persone affette da questa grave forma di distrofia muscolare.
Il Rally di Alba, secondo appuntamento del Trofeo Rally Asfalto dell'anno, richiama sempre un grande pubblico perchè, come è ormai tradizione, regala agli appassionati dei grandi contenuti sportivi e sottolinea le attitudini culturali e turistiche della rinomata zona della Langa cuneese.
I volontari di Parent Project saranno presenti sabato 5 aprile, dalle ore 16 alle ore 22 e domenica 6 aprile, dalle ore 9 alle ore 15 in piazza Medford, nei pressi della Direzione gara del rally, con uno stand presso il quale sarà possibile ricevere informazioni sulla patologia ed effettuare le donazioni.
La Distrofia Muscolare di Duchenne e Becker è una malattia rara, la forma più grave delle distrofie muscolari, che colpisce 1 su 3.500 maschi nati vivi. Si stima che in Italia ci siano 5.000 persone affette dalla patologia. Attualmente non esiste una cura specifica, ma un trattamento da parte di una equipe multidisciplinare che ha permesso di migliorare le condizioni generali e raddoppiare le aspettative di vita.
Parent Project Onlus, nasce nel 1996 per sconfiggere questa grave distrofia muscolare promuovendo e finanziando la ricerca scientifica. In questi anni ha contribuito, con oltre 1.500.000 euro di investimenti, al finanziamento di più di 50 progetti specifici. Altro obiettivo primario dell'associazione è la formazione e il sostegno, per le famiglie coinvolte dalla malattia. Dal 2002, grazie al Centro Ascolto Duchenne, finanziato dall'ISMA (Istituti Santa Maria in Aquiro di Roma) e dalla Fondazione Peppino Vismara, è attivo un servizio gratuito che fornisce informazioni del quale possono beneficiare, oltre alle famiglie, tutti gli specialisti interessati all'approfondimento.
Maggiori informazioni, sulle attività di Parent Project e del Centro Ascolto Duchenne è possibile riceverle telefonando al Numero Verde 800 - 943 333 o visitando il sito internet www.parentproject.org
Per sostenere i progetti di Parent Project Onlus c/c postale 94255007
Banca Credito Cooperativo Ag. 19 IBAN IT 38 V 08327 03219 000000005775
Ufficio stampa Parent Project Onlus
Stefania Collet
Tel. 06 66.18.28.11
Fax 06 66.18.84.28
e-mail ufficiostampa@parentproject.org
www.parentproject.org
Convegno di Milano: terapia farmacologica
In realtà, sotto il termine di “terapia farmacologica” sono raggruppati tutta una serie di approcci diversi che hanno come obiettivo finale quello di sviluppare delle molecole - tra cui moderni farmaci biotech - che possano agire direttamente sul danno a livello del gene (PTC124), regolare i meccanismi responsabili della rigenerazione del tessuto muscolare (ACE031) o, più semplicemente, cercare di rendere più forti i muscoli anche in mancanza di distrofina.
Steroidi giornalieri: una strategia valida a lungo termine?
 Douglas Biggar
Douglas Biggar
Nonostante gli enormi progressi fatti negli ultimi anni nel campo della DMD non esiste ancora nessun trattamento terapeutico veramente efficace per arrestare il lento avanzamento della patologia. In questo panorama, i farmaci corticosteroidi (cortisonici) sembrano avere un ruolo rilevante, intervenendo sui processi anti-infiammatori e riducendo le reazioni immunitarie coinvolte nella progressione della malattia. È importante chiarire che si tratta comunque di una terapia palliativa e non di un trattamento in grado di risolvere il problema ma, indipendentemente dall’esatto meccanismo d’azione che è ancora sconosciuto e in virtù degli effetti positivi riscontrati, i cortisonici sono ampiamente utilizzati nella distrofia di Duchenne.
Nei pazienti Duchenne i corticosteroidi esercitano un’azione anabolica sul muscolo, aumentando cioè la massa muscolare, e riducono i fenomeni degenerativi, intervenendo cosi favorevolmente sulla forza muscolare e sul decorso clinico. Se presi prima della perdita della capacita’ di camminare, i corticosteroidi sono in grado di prolungare la deambulazione di qualche anno.
A fronte di una certa efficacia l’utilizzo cronico di questi farmaci è purtroppo limitato da una serie di effetti collaterali rilevanti, quali l’arresto della crescita, un notevole aumento del peso, aumento della glicemia, osteoporosi, cataratta e un cambiamento del comportamento che può sviluppare una indole aggressiva. Per questi motivi, uno dei punti piu’ discussi dalla comunità medico-scientifica è la valutazione del rapporto rischi–benefici della terapia cortico-steroidea.
Al convegno Douglas Biggar, professore all'Università di Toronto in Canda, ha illustrato i risultati ottenuti da studi basati sul trattamento prolungato (da 10 a 20 anni) con il Deflazacort, un derivato più tollerabile del Prednisone con pari efficacia nel migliorare la forza e la funzionalità del muscolo e nel prolungare la capacità di camminare. E' stato osservato che in alcuni pazienti il trattamento porta a dei benefici a lungo termine su diversi importanti aspetti come la deambulazione, la funzione cardiaca e respiratoria, la forza muscolare e la scoliosi. Gli effetti collaterali risultano sempre presenti ma più accettabili: l’aumento di peso può essere mantenuto sotto controllo con una semplice dieta, l’osteoporosi non sembra rilevante e può essere trattata con assunzioni giornaliere di vitamina D e calcio, l’insorgenza di cataratta non compromette la vista e non necessita di trattamenti specifici, inoltre non si è manifestata ipertensione o aumento di glicemia.
Gli studi effettuati mostrano che il Deflazacort può avere un importante impatto sul decorso della distrofia di Duchenne e sulla qualità di vita dei pazienti, una somministrazione prolungata rallenta notevolmente la degenerazione muscolare, riducendo le cure cliniche necessarie dopo i 10 anni di vita. Rimane il fatto che per avere un panorama più completo della situazione sono ancora necessarie ulteriori analisi, più approfondite e prolungate nel tempo, progetto al quale stanno lavorando i ricercatori canadesi.
Aggiornamenti sullo sviluppo del PTC124 per la DMD
 Langdon Miller
Langdon MillerImmaginiamo di paragonare la struttura di un gene a quella di una frase: per essere letto un gene ha bisogno di un segnale di inizio e di uno di stop, proprio come usiamo fare con la maiuscola ed il punto finale in una frase. Tra i diversi tipi di mutazioni che possono colpire i nostri geni vi è il codone di stop prematuro, si tratta di una mutazione puntiforme, chiamata “non senso”, che comporta l’introduzione di un segnale di stop in una zona interna del gene. Questa mutazione causa l’interruzione anticipata della lettura del gene e, quindi, la produzione di una forma più corta, non funzionale, della distrofina. Per riprendere il nostro paragone iniziale: è come se si introducesse un punto nel bel mezzo di una frase, si perderebbe parte dell’informazione ed il senso stesso della frase.
Circa il 10-15% dei ragazzi colpiti da distrofia muscolare di Duchenne hanno un codone di stop prematuro nel gene della distrofina. Da alcuni anni i ricercatori della PTC Therapeutics, una società biofarmaceutica americana specializzata nella scoperta e nello sviluppo di piccole molecole farmacologiche che intervengono sui meccanismi molecolare post-trascrizionali (ovvero quei meccanismi coinvolti nella lettura dei geni e nella loro traduzione in proteine), hanno messo a punto un nuovo tipo di farmaco sperimentale che permetta al macchinario cellulare di ignorare il segnale di stop e di continuare così la corretta lettura del gene. Si tratta del PTC124, un farmaco per uso orale già designato dall’FDA come “orphan drug” per il trattamento della Fibrosi Cistica e della DMD causate da mutazioni “non senso”.
Negli ultimi anni, la ricerca focalizzata sul possibile uso clinico del PTC124 ha dato risultati molto incoraggianti che hanno permesso di passare dagli studi preliminari effettuati in laboratorio su colture cellulari e su animali, alle prime sperimentazioni cliniche di fase I e II sull’uomo.
I risultati degli esperimenti pre-clinici sono stati pubblicati a maggio 2007 sulla rivista Nature. Gli studi in linee cellulari con mutazioni “non senso” della distrofina hanno indicato che il PTC124 può ripristinare la produzione della distrofina mancante. Nel topo mdx (il modello murino della DMD causata da una mutazione non senso), una somministrazione orale del PTC124 della durata di 4 settimane ha indotto la produzione di distrofina nei muscoli scheletrici e nel diaframma associata ad una riduzione della fragilità muscolare.
Questi dati hanno promosso lo studio del PTC124 alla sperimentazione clinica di fase I e successivamente a quella di fase IIa. I risultati, ottenuti verso la fine del 2007, hanno dimostrato che l’assunzione di PTC124 induce in modo sicuro un ripristino parziale della produzione della distrofina completa e riduce la fragilità muscolare. Attività e sicurezza sono state inoltre confermate in pazienti affetti da fibrosi cistica causata da mutazione “non senso”, che hanno assunto il farmaco per un massimo di circa 3 mesi.
Dopo aver illustrato le caratteristiche e i risultati degli studi basati sul PTC124, Langdon Miller - Direttore clinico della PTC Therapeutics - ha annunciato l’avvio di un nuovo trial clinico internazionale che includerà anche centri italiani. L’obiettivo primario dello studio è di valutare l’effetto del PTC124 sulla capacità di deambulazione di ragazzi con la distrofia muscolare Duchenne o Becker, causata da mutazione “non senso”. Si tratta di uno studio di fase IIb, multicentrico(che si svolge in più di una struttura sanitaria), randomizzato (scelta casuali dei diversi gruppi di ragazzi per le diverse somministrazioni), dose-ranging (che prevede l’utilizzo di frazioni differenti della dose prevista per avere un effetto farmacologico) e in doppio cieco (protocollo sperimentale per cui né chi somministra, né chi assume il farmaco, sia a conoscenza della vera natura del trattamento in esame) controllato con placebo.
L’arruolamento previsto dalla PTC Therapeutics è di circa 165 pazienti, in 25 centri di sperimentazione in tutto il mondo tra cui compaiono anche 3 centri italiani: il Policlinico Universitario "Agostino Gemelli" di Roma (Dott. Eugenio Mercuri), l’Ospedale pediatrico Bambin Gesù (Dott. Bertini) e l’Ospedale Maggiore Policlinico di Milano.
Per essere inclusi nel trial, i pazienti devono avere le seguenti caratteristiche: essere di sesso maschile e avere dai 5 anni in sù; essere in possesso di una diagnosi genetica che indichi la presenza di una mutazione “non senso” puntiforme nel gene della distrofina; avere la capacità di camminare per 75 metri continuativamente in 6 minuti (definita come Distanza Percorsa in 6 Minuti 6MWD), senza assistenza né tutori ortopedici.
L’inizio dell’arruolamento è previsto per i prossimi mesi, mentre la sperimentazione clinica dovrà iniziare entro gennaio 2009 e finire entro gennaio 2010, il trattamento ha la durata di 48 settimane.
Gli inibitori delle deacetilasi
 Pierlorenzo Puri
Pierlorenzo PuriNormalmente, la rigenerazione - o “riparazione” - muscolare avviene ad opera delle cellule satellite. Quando il muscolo scheletrico subisce un danno, alcuni segnali cellulari vanno a “risvegliare” ed attivare queste cellule, le quali proliferano, si differenziano e si fondono con le fibre muscolari. Uno degli eventi chiave del processo differenziativo è il rimodellamento della cromatina delle cellule. La cromatina è l’impalcatura dell’informazione genetica ed è formata da Dna e proteine: è una struttura altamente dinamica che si divide in regioni più lasse, in cui vi è l’attivazione dei geni, e regioni più compatte, in cui i geni sono mantenuti silenti. In una cellula la scelta di quali regioni della cromatina mantenere lasse o compatte, e cioè quali geni accendere o spengere, ne definisce l’identità. Con il rimodellamento della cromatina le cellule satellite passano da uno stato di cellule staminali quiescenti ad uno stato di cellule muscolari differenziate, esprimenti i geni specifici del muscolo, che vanno ad integrarsi e a riparare il muscolo danneggiato.
I segnali cellulari che risvegliano le cellule satellite sono diversi e possono essere innescati da proteine del processo infiammatorio o da fattori ormonali (ad esempio il testosterone, l’insulina, IGF1), entrambi attivati dopo un danno muscolare.
Negli ultimi anni, le ricerche guidate da Pier Lorenzo Puri hanno permesso la caratterizzazione di alcuni meccanismi molecolari con i quali i segnali cellulari vengono convertiti in rimodellamento della cromatina, e successiva accensione del set specifico di geni. Con una serie di lavori pubblicati tra il 1997 ed il 2002, Puri ha dimostrato come le acetiltrasferasi, proteine che modificano la struttura chimica degli istoni (i “lucchetti” che avvolgono il Dna), siano determinanti per il differenziamento muscolare. Queste acetiltrasferasi, in particolare p300 e PCAF, collaborano con un’altra proteina, MyoD, nota per essere il fattore che istruisce i progenitori muscolari (ad esempio le cellule satelliti) a riparare il muscolo distrofico. L’azione viene svolta a livello della cromatina: quest’ultima acquisisce una struttura più lassa, in punti specifici chiamati loci muscolari, con la conseguente attivazione dei geni che conferiscono l’identità muscolare.
A controbilanciare l’azione delle acetiltrasferasi sono le deacetilasi: proteine che, sempre agendo sugli istoni, richiudono invece la cromatina in uno stato più compatto e silente. In uno studio, pubblicato a settembre 2006 su Nature Medicine, il gruppo di ricerca di Puri ha dimostrato che l’utilizzo di un inibitore delle deacetilasi (la Tricostatina A) è in grado di arrestare la progressione della degenerazione muscolare e di promuovere la rigenerazione del muscolo scheletrico in topi mdx. Le analisi effettuate hanno inoltre evidenziato che il trattamento con Tricostatina A da una parte determina un aumento della massa muscolare, e d’altra parte preserva i muscoli dalla perdita di forza e dalle alterazioni morfologiche tipiche. Dopo soltanto 3 mesi di trattamento i topolini malati sono riusciti infatti a correre normalmente.
In particolare, i ricercatori hanno evidenziato che l’effetto della Tricostatina A passa attraverso l’azione della follistatina, una proteina prodotta dalle cellule del muscolo scheletrico che inibisce la funzione della miostatina. La miostatina è, a sua volta, una proteina inibitrice che interagisce negativamente con lo sviluppo muscolare. Negli individui adulti l`azione contrapposta di miostatina (che limita la rigenerazione muscolare) e follistatina (che induce invece la rigenerazione muscolare) appare regolare la crescita della massa muscolare. Queste evidenze suggeriscono che l'antagonismo funzionale tra miostatina e follistatina possa costituire un importante bersaglio farmacologico per la terapia di distrofie muscolari.
In quest’ottica, definire i meccanismi biochimici e molecolari con cui gli inibitori delle deacetilasi istruiscono la cromatina a modulare l’espressione dei geni coinvolti nella rigenerazione muscolare rappresenta un punto fondamentale per le future ricerche.
Ossido Nitrico e agenti anti-infiammatori non steroidei
 Emilio Clementi
Emilio ClementiL’ossido Nitrico (NO) è normalmente prodotto dai muscoli scheletrici ed ha effetti benefici sulla funzione muscolare. Il meccanismo di azione del NO si basa sulla stimolazione di fattori chiave coinvolti nel processo di rigenerazione delle cellule muscolari, ad esempio l’attivazione delle cellule satellite (le cellule staminali adulte del tessuto muscolare) o il rilascio di fattori miotrofici, ovvero che inducono la crescita della massa muscolare. Inoltre, l’NO stimola la vasodilatazione ed il conseguente aumento di apporto di ossigeno e glucosio alle cellule, fattori che contribuiscono a proteggere i muscoli dai danni a seguito di contrazioni.
Uno studio guidato da Emilio Clementi, e pubblicato nel novembre 2006 sulla rivista PNAS, ha dimostrato come la combinazione di farmaci che rilasciano NO con anti-infiammatori non steroidei (NSAID) abbia effetti positivi sul riparo del tessuto muscolare. Una sperimentazione pre-clinica, della durata di un anno, e’ stata effettuata su topi modello per la distrofia dei cingoli e la DMD mediante somministrazione orale di anti-infiammatori non steroidei che rilasciano NO (indicati con la sigla NO-NSAID).
Emilio Clementi, ricercatore al San Raffaele di Milano, ha presentato lo studio in cui sono stati usati due farmaci distinti, ma con caratteristiche similari: il nitro-ibuprofene e il nitro-paracetamolo. I risultati del trattamento sui topi hanno mostrato un significativo miglioramento degli aspetti morfologici, biochimici e funzionali dei muscoli, senza la comparsa di effetti collaterali di rilevanza. Le analisi effettuate dal team italiano hanno evidenziato che i farmaci rallentano la progressione della distrofia agendo su vari fronti: riducono il processo infiammatorio, prevengono il danno muscolare e mantengono il numero e la funzionalità delle cellule satellite. Studi comparativi hanno inoltre dimostrato che i NO-NSAID sono significativamente più efficaci dei corticosteroidi.
Questi farmaci hanno anche una sorprendente influenza positiva sugli effetti terapeutici dei mesoangioblasti iniettati per via sistemica. In particolare i NO-NSAID aumentano le capacitaà migratorie e di integrazione nel tessuto muscolare che hanno queste cellule staminali.
I NO-NSAID sono farmaci già autorizzati per l’uso clinico sull’uomo, non presentano gli effetti collaterali che danno i corticosteroidi e non provocano nessun tipo di danno a livello gastrointestinale. La speranza è di poter avviare al più presto la sperimentazione clinica per la DMD.
Aumentare la massa muscolare con ACE 031
 Jas Seehra
Jas SeehraLa miostatina, anche conosciuta come GDF-8, è una proteina prodotta dalle cellule del muscolo scheletrico che interagisce negativamente con lo sviluppo muscolare, ovvero limita la crescita della massa muscolare.
In passato è stato osservato che in animali, ma anche nell’uomo, in cui la funzionalità della miostatina viene a mancare a causa di mutazioni a carico del gene corrispondente, si ha un aumento della massa e della forza muscolare. Viceversa, una produzione esagerata della proteina porta ad una perdita del tessuto muscolare accompagnata da una diminuzione della forza dei muscoli. Partendo da queste osservazioni, numerosi studi hanno successivamente evidenziato come un blocco della funzione della miostatina possa avere un effetto terapeutico sulla DMD.
Per agire sul tessuto muscolare la miostatina deve legarsi ad un recettore specifico posto sulla superficie delle cellule muscolari. Negli ultimi anni, la società farmaceutica Americana Acceleron ha sviluppato un analogo di questo recettore, chiamato ACE 031, in forma iniettabile. Questa molecola terapeutica iniettata nei muscoli ha una funzione antagonista, cioè lega la miostatina sequestrandola dai recettori naturali che, quindi, non risentono più dell’effetto inibitorio della proteina.
La sperimentazione pre-clinica, effettuata in diversi modelli animali (dai topi alle scimmie), ha dimostrato che l’utilizzo di ACE 031 porta ad un incremento della massa muscolare, lo stesso è stato dimostrato in topi mdx, il che fornisce speranza per lo sviluppo di una terapia per la DMD.
Francesca Ceradini
INCONTRO CON IL RETTORE A ROMA
Il 18 marzo 2008, presso la prima università di Roma, si è svolto un incontro in cui è stato presentato al Rettore Guarini il primo "spin-off sociale" per la distrofia muscolare di Duchenne.
Un modello di impresa che ha lo scopo di trasferire la conoscenza tecnologica e reinvestire gli utili nella ricerca scientifica. Il progetto coinvolge: il Consorzio Sapienza ed Innovazione, il Dipartimento di Genetica e Biologia Molecolare della Sapienza e Parent Project onlus.
Il Consorzio, si sta occupando di elaborare uno spin-off che consenta di arrivare a produrre una cura per la distrofia di Duchenne. Questa operazione permetterà all'Università di veder finalmente riconosciute le prestigiose attività scientifiche condotte dai suoi ricercatori e alle Aziende di acquisire il know-how universitario necessario per dedicarsi direttamente allo sviluppo produttivo e alla commercializzazione delle invenzioni.
 Si tratta di un'operazione dal valore sociale innovativo che, come è gia avvenuto in Olanda e negli Stati Uniti, si spera consentirà alle associazioni di recuperare risorse economiche determinanti per proseguire con le ricerche.
Si tratta di un'operazione dal valore sociale innovativo che, come è gia avvenuto in Olanda e negli Stati Uniti, si spera consentirà alle associazioni di recuperare risorse economiche determinanti per proseguire con le ricerche.
Il ruolo di Parent Projectsarà quello di garantire il rispetto delle urgenze e delle necessità dei pazienti.
Nel corso dell'evento sono stati anche illustrati gli sviluppi del brevetto nato dalla Prof.ssa Irene Bozzoni, del Dipartimento di Genetica e Biologia Molecolare, che utilizza la tecnica dell'Exon skipping,attualmente in fase di studio preclinico e che sarà oggetto dello spin off.
vai al comunicato stampa
Exon skipping in Italia
COMUNICATO 18.03.2008
L’Università La Sapienza di Roma e Parent Project per un modello di impresa che consentirà di trasferire la conoscenza tecnologica e reinvestire gli utili nella ricerca scientifica.
Presentato al Rettore Guarini il primo “spin-off sociale” per la distrofia muscolare di Duchenne.
“Questo incontro rappresenta, prima di tutto, la volontà del mondo accademico di iniziare a lavorare anche al fianco delle associazioni di pazienti per valorizzare le acquisizioni scientifiche e trasformarle in reali opportunità di cambiamento.” – ha dichiarato Filippo Buccella, presidente di Parent Project onlus in occasione dell’incontro con il rettore Renato Guarini durante il quale sono stati illustrati i promettenti sviluppi di un brevetto nato dalla Prof.ssa Irene Bozzoni, del Dipartimento di Genetica e Biologia Molecolare, che utilizza l’Exon skipping ed è attualmente in fase di studio preclinico.“Il Consorzio Sapienza Innovazione – per il quale erano presenti il presidente Prof. Luciano Caglioti, prorettore per la ricerca, lo sviluppo e i contatti con il mondo produttivo, e il direttore Dr. Stephen Trueman - ha svolto un lavoro straordinario per l’avvio di questo spin-off, che per la prima volta avrà un valore sociale innovativo e, come è gia avvenuto in Olanda e negli Stati Uniti, consentirà alle associazioni di recuperare risorse economiche determinanti per proseguire con le ricerche.” – ha ricordato il presidente di Parent Project -
“Lo spin-off che stiamo elaborando per arrivare a produrre una cura per la distrofia di Duchenne, è un modello di efficienza ed innovazione che permetterà all’Università di veder finalmente riconosciute le prestigiose attività scientifiche condotte dai ricercatori e alle Aziende di acquisire il know-how universitario per dedicarsi direttamente allo sviluppo produttivo e alla commercializzazione delle invenzioni.” – hanno sottolineato il Prof. Luigi Frati, Rettore Vicario e Preside della Facoltà di Medicina e Chirurgia e il Prof. Renzo Piva, Presidente della Commissione Innovazione della ricerca e della tecnologia
“Il ruolo di Parent Project - conclude Buccella – sarà quello di garantire il rispetto delle urgenze e delle necessità dei pazienti.”
La Distrofia Muscolare di Duchenne e Becker (DMD/DMB) è una malattia rara, la forma più grave delle distrofie muscolari, che viene trasmessa dalle madri attraverso il cromosoma X e colpisce 1 su 3.500 maschi nati vivi. Attualmente non esiste una cura specifica, ma un trattamento da parte di una equipe multidisciplinare che ha permesso di migliorare le condizioni generali e raddoppiare le aspettative di vita. Si stima che in Italia ci siano 5.000 persone affette dalla patologia.
Il Parent Project Onlus, nasce nel 1996 per sconfiggere questa grave distrofia muscolare promuovendo e finanziando la ricerca scientifica. In questi anni ha contribuito, con oltre 1.500.000 euro di investimenti, al finanziamento di più di 50 progetti specifici tra i quali lo studio di Irene Bozzoni, con la tecnica dell’exon skipping con antisenso chimerici; Pier Lorenzo Puri, del Burnham Istitute di La Jolla – California, basato sull’applicazione di alcuni agenti farmacologici chiamati inibitori delle deacetilasi e Giulio Cossu, dell’Istituto San Raffaele di Milano, con le cellule staminali. Altro obiettivo primario dell’associazione è la formazione e il sostegno, per le famiglie coinvolte dalla malattia.
Maggiori informazioni sulle attività di Parent Project al Numero Verde 800 943 333 o sul sito www.parentproject.it
Ufficio stampa Parent Project Onlus
Tel. 06 66.18.28.11
Cell. 349 5737747
Fax 06 66.18.84.28
Stefania Collet
e-mail ufficiostampa@parentproject.org
CONVEGNO DI MILANO: TERAPIA GENICA
![]() Terapia genica ed Exon skipping
Terapia genica ed Exon skipping
Continuiamo con il resoconto del convegno “I trial clinici e la ricerca Duchenne e Becker nel mondo” dello scorso febbraio a MIlano. Questa parte è dedicata alla terapia genica ed in particolar modo all'exon skipping.
Nell’ottica di una terapia genica per la DMD, l’ostacolo più importanteè rappresentato dalle enormi dimensioni del gene della Distrofina: 2.4Mb, il più grande gene contenuto nel nostro DNA. Ciò fa si che la normale sostituzione genica non si presenti come un approccio facile,ed è per questo che nel corso degli anni sono state studiate tutta un aserie di strategie alternative. Dalla creazione di vettori adatti al trasporto dell'intero gene della distrofina, alla creazione di geni sostitutivi più corti(minidistrofina), all'approccio dell’exon skipping.
Alcuni di questestrategie sono state presentate da ricercatori venuti da diversi angoli del mondo.
Terapia genica con vettori AAV
 Annick Martin
Annick Martin
Con la sua presentazione, Annick Martindell'Associazione Francese contro le Miopatie (AFM), associazione creata nel 1958 da un gruppo di famiglie di pazienti per sostenere lo sviluppo di nuovi strumenti scientifici per lo studio di malattie genetiche rare, ha illustrato alcuni progetti di ricerca in corso che si basano sull'utilizzo diVirus Adeno-Associati (AAV) come "mezzo" per veicolare geni nelle cellule.
Questi vettori hanno il vantaggio di avere una buona efficienza di trasferimento nelle cellule muscolari e cardiache, mantenendo unarisposta immunitaria ridotta. Inoltre non c’è integrazione del gene introdotto nel DNA delle cellule ospite, e ciò elimina la possibilità di insorgenza tumorale. Per di più l’espressione del gene introdotto viene mantenuta per anni, in maniera stabile, nelle cellule trattate. Negli ultimi anni, gli AAV sono stati i vettori scelti per la maggior parte delle sperimentazioni cliniche. Ad oggi, nel mondo, sono in corso ben 28 trial clinici con AAV con dati positivi circa la loro efficacia.
Un limite dei vettori AAV è però la loro dimensione, sono piuttosto piccoli e non possono veicolare materiale genetico superiore alle 5mila basi di lunghezza. La parte codificante del gene della distrofina che con i suoi 79 esoni è lunga circa 14mila basi deve essere quindi accorciata per poter essere inserita in un AAV. Negli ultimi anni sono stati ideati studi con l’utilizzo di una forma più corta del gene della distrofina, chiamata minidistrofina, “costruita” in maniera tale che le regioni fondamentali per il funzionamento della proteina vengano mantenute. Un approccio che non prevede una vera e propria cura, piuttosto una conversione della distrofia muscolare di Duchenne nella forma più lieve di tipo Becker.
Ad oggi sono in corso diversi studi basati sul sistema AAV-minidistrofina, tra cui quello portato avanti dalla biotech Askbio che ha avviato una sperimentazione clinica su di una forma corta di distrofina chiamata Biostrophin. Per il momento lo studio sta valutando la sicurezza dell’approccio terapeutico mediante la somministrazione intramuscolare in sei ragazzi DMD.
Un altro filone, molto promettente, è l’utilizzo di AAV per veicolare all’interno delle cellule muscolari un gene terapeutico. Questo gene (parliamo di geni diversi a seconda del gruppo di ricerca) codifica per molecole di RNA disegnate ad hoc per effettuare l’exon skipping. Su questa ricerca sono impegnati un gruppo francese ed un gruppo italiano (vedere più avanti).
Altri approcci di terapia genica, in fase ancora molto preliminare, sono invece basati sulla modulazione del gene per l’utrofina o la somministrazione di agenti anti-miostatina.
L'exon skipping
L’exon skipping (letteralmente tradotto come “salto dell’esone”) è una tecnica che mira ad eliminare il “danno molecolare” modificando direttamente l’RNA messaggero che codifica per la distrofina.
La terapia basata sull’exon skipping non può essere considerata una cura vera e propria, piuttosto un modo per convertire la distrofia di Duchenne in quella di tipo Becker, ovvero un modo per ridurre la gravità della distrofia. Quando una mutazione cambia lo schema di lettura del gene della distrofina, chiamato in gergo “reading frame”, non vi è più la produzione della proteina funzionale e ciò causa l’insorgenza della DMD. Il corretto schema di lettura del gene può essere ristabilito eliminando direttamente uno o più esoni corrispondenti alla regione in cui è presente la mutazione.
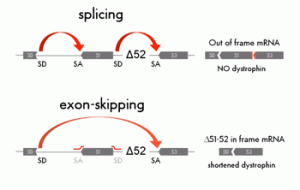 Questo processo di eliminazione viene effettuato usando dei corti frammenti di RNA, chiamati antisenso, che si appaiano in zone specifiche dell’RNA messaggero (in particolare alle giunzioni di splicing degli esoni). Alla fine di questa “operazione molecolare”, la distrofina prodotta sarà più corta del normale ma, se il pezzo eliminato non corrisponde ad una regione cruciale, la proteina potrà ancora svolgere la sua funzione muscolare. La versione corta presenta dei difetti, ma una distrofina meno funzionale è pur sempre meglio rispetto alla quasi completa assenza. Insomma, il concetto è di limitare i danni agendo direttamente sulla causa.
Questo processo di eliminazione viene effettuato usando dei corti frammenti di RNA, chiamati antisenso, che si appaiano in zone specifiche dell’RNA messaggero (in particolare alle giunzioni di splicing degli esoni). Alla fine di questa “operazione molecolare”, la distrofina prodotta sarà più corta del normale ma, se il pezzo eliminato non corrisponde ad una regione cruciale, la proteina potrà ancora svolgere la sua funzione muscolare. La versione corta presenta dei difetti, ma una distrofina meno funzionale è pur sempre meglio rispetto alla quasi completa assenza. Insomma, il concetto è di limitare i danni agendo direttamente sulla causa.L’exon skipping rappresenta una delle tecniche più promettenti del momento e su di essa si stanno impegnando diversi gruppi di ricerca nel mondo. Da una parte vi è l'exon skipping mediato da semplici molecole di RNA (l’approccio farmacologico), e dall’altra l’exon skipping effettuato mediante tecniche di terapia genica. Negli ultimi due anni si è sentito molto parlare dell’avvio di trial clinici con
AON (Oligonucleotidi antisenso), ovvero molecole di RNA sintetizzate in laboratorio, su cui stanno puntando Prosensa, un’azienda biotech olandese, e MDEX, un consorzio britannico. L’altro filone di ricerca, portato avanti da un gruppo italiano ed uno francese, si basa invece sull’utilizzo di AAV (Virus Adeno-Associati) per veicolare i “geni terapeutici” che codificano per l’RNA necessario all’exon skipping. Gli studi sono prevalentemente focalizzati sull’eliminazione dell’esone 51, questo perche’ il salto di questo esone permette di agire sull’effetto delle Delezione degli esoni 48, 49 e 50 che sono le piu’ frequenti.Gli AON in uso in questo momento sono di due tipi: da una parte ci sono i2-O-methyl AON, sperimentati dagli olandesi, e dall’altra parta i
Morpholino, sperimentati dagli inglesi e dagli australiani. Entrambi gli AON sono oligonucleotidi chimicamente modificati, ciò li rende più stabili e non vengono distrutti all’interno delle cellule muscolari. Alcuni aspetti legati all’efficacia di azione in determinate condizioni li differenzia ma i due approcci sembrano, al momento, essere entrambi promettenti.1- Gli AON olandesi
 Annemieke Aartsma-Rus
Annemieke Aartsma-RusProsensa è una company biotech olandese creata 5 anni fa da ricercatori universitari. Gli olandesi hanno il merito di aver avviato, nel 2006, il primo studio clinico basato sull’uso di AON su pazienti DMD. Alla conferenzaAnnemieke Aartsma-Rus, dell'Università di Leiden in Olanda, ha presentato lo stato dell'arte della tecnica e i risultati sulle sperimentazioni cliniche concluse ed in corso.
Le prime sperimentazioni di fase II sono state effettuate su 4 ragazzi DMD, di età compresa tra gli 8 ed i 16 anni, con singole iniezioni di un 2-O-methyl AON (PRO 051) contro l’esone 51 nel muscolo tibiale anteriore. I risultati sono davvero incoraggianti: analisi molecolari e istologiche su biopsie del muscolo hanno mostrato una produzione di nuova distrofina ad un mese dall’iniezione. Lo scopo del primo studio è stato di provare che l’exon skipping dell’esone 51 funzioni per diversi tipi di mutazioni, infatti i ragazzi reclutati presentano delezioni su diversi esoni adiacenti al 51. Si puo’ dire quindi che l’ipotesi formulata funziona.
Ora il team olandese ha avviato una nuova sperimentazione: la somministrazione degli AON per
via sistemica. Questa seconda via permetterebbe agli AON di raggiungere i vari distretti del corpo ed agire così su tutti i muscoli colpiti dalla distrofia. Diversi oligonucleotidi sono già stati testati, e messi a punto, con esperimenti condotti su topi e scimmie. Dai risultati preliminari i ricercatori hanno deciso di focalizzare il trial clinico su 2 diversi AON, uno contro l’esone 51 e l’altro contro l’esone 46. E’ previsto che lo studio sia condotto in Europa e negli Stati Uniti, su un numero compreso tra i 15 e i 20 pazienti DMD. Questi verranno trattati con una iniezione settimanale per 5 settimane di seguito e, oltre alle analisi molecolari sulla distrofina, verranno analizzate la forza e la funzionalità muscolare dei ragazzi.2- I morpholino inglesi
Dopo gli olandesi è stata la volta degli inglesi: il 22 ottobre 2007 il consorzio MDEX ha avuto l’approvazione da parte delle autorità britanniche di iniziare un trial clinico di exon skipping, su pazienti DMD, basato sull’uso di morpholino.
Il trial clinico, avviato sul primo ragazzo lo scorso dicembre, sara’ effettuato su 9 ragazzi DMD di età compresa tra i 13 e i 17 anni. I pazienti saranno divisi in 3 gruppi per la somministrazione di 3 dosi diverse di morpholino, le dosi verranno iniettato localmente in un piccolo muscolo del piede (extensor digitorum brevis) mediante un’unica somministrazione. Le analisi molecolari e istologiche su biopsie di muscolo saranno effettuate dopo 21 giorni dal trattamento. E’ stato scelto il muscolo del piede perché non è, in alcun modo, un muscolo essenziale e si può facilmente rimuovere in caso di eventuali effetti secondari nocivi.
Se i primi risultati saranno positivi, gli inglesi sono intenzionati ad iniziare, il piu’ velocemente possibile, una seconda fase della sperimentazione con la somministrazione sistemica del morpholino. Lo scopo di questo successivo studio sarà, come per gli olandesi, far arrivare la “molecola terapeutica” in tutti i muscoli del corpo.
3- AON per tutti
 Steve Wilton
Steve WiltonSteve Wilton, professore all'University of Western Australia, è uno dei pionieri dell’exon skipping, la sua idea di utilizzare questa metodologia per studiare i meccanismi molecolari sulla distrofina risale al 1996. Nel corso degli anni il ricercatore australiano ha studiato diverse strategie e messo a punto diversi AON (sia 2-O-methyl che morpholino) per una possibile terapia contro la DMD.
Il gruppo di ricerca guidato da Wilton ha effettuato uno screening di AONsu circa cinquecento 2-O-methyl oligonucleotidi disegnati per tutti gli esoni della distrofina, con risultati molto interessanti. “Sono felice di poter affermare che nel mio laboratorio siamo oramai in grado di effettuare l’exon skipping di ogni esone presente nel gene della distrofina – ha dichiarato Wilton - più precisamente dall’esone 2 all’esone 78, visto che il primo e l’ultimo esone sono essenziali e non eliminabili”. Gli esperimenti, pubblicati a febbraio 2007 su Molecular Therapy, sono stati effettuati in colture di cellule muscolari: tutti e 77 gli esoni bersaglio sono stati eliminati con successo. L’efficienza tuttavia non è la stessa per tutti gli AON testati, alcuni funzionano meglio, questo avviene perché certi esoni sono più facilmente rimovibili di altri per i quali, ad esempio, è necessario utilizzare un “cocktail” di diversi AON.
In questi ultimi mesi, i ricercatori stanno mettendo a punto nuove strategie per rendere più efficiente l’exon skipping di questi esoni “refrattari”. Lo scopo è quello di ottenere un certo numero di oligo, che siano buoni candidati per ogni tipo di esone, mediante i quali avviare nuove sperimentazioni sui
modelli animali e, il prima possibile, sui pazienti.4- L'exon skipping italiano
 Fernanda De Angelis
Fernanda De AngelisI primi importanti risultati del gruppo di Irene Bozzoni,pubblicati nel 2002 sulla rivista scientifica PNAS (Proceeding of the National Academy of Sciences), sono stati ottenuti su colture di cellule muscolari (i
mioblasti) di un paziente DMD con una delezione degli esoni 48, 49 e 50. In questo studio sono stati iniettati nei mioblasti alcuni vettori retroviralicontenenti “geni terapeutici” codificanti RNA antisenso in grado di riconoscere le giunzioni di splicing specifiche per eliminare l’esone 51. Con questa strategia è stato dimostrato, prima di tutto, che le molecole di RNA prodotte nelle cellule sono stabili e la loro azione è continuativa nel tempo e, successivamente, che si ha il ripristino della produzione di distrofina, che anche se più corta, è funzionante.Vista l’efficacia della strategia messa a punto, lo studio è stato esteso al modello animale per la DMD: il topo
mdx. Questo topo ha una mutazione puntiforme “non senso”, ovvero la formazione di un codone di stop, nell’esone 23 del gene per la distrofina, con la conseguente produzione di una distrofina non funzionale. In questo caso come “navetta” di trasporto per i geni terapeutici sono stati utilizzati AAV. Al convegno Fernanda De Angelis, ricercatrice presso il gruppo di Irene Bozzoni, ha presentato i dati sperimentali dello stusio. La somministrazione locale (per via intramuscolare) o in tutto il corpo (pervia sistemica, mediante la vena nella coda del topo) ha dato risultati molto positivi che sono stati pubblicati su PNAS nel 2006. Le analisi molecolari hanno dimostrato la presenza di alti livelli di RNA antisenso nei muscoli del topo, i quali hanno indotto l’exon skipping dell’esone 23 determinando la produzione di distrofina e, soprattutto, il recupero della funzionalità muscolare. L’introduzione di virus AAV per via sistemica ha permesso di ottenere un’efficiente trasporto degli RNA in tutti i distretti muscolari, inclusi il cuore e il diaframma che rappresentano i tessuti più importanti per il trattamento terapeutico dei malati DMD.Successivi esperimenti effettuati sui topi mdx hanno mostrato l’effetto a lungo termine degli RNA antisenso: 18 mesi dopo l’iniezione sistemica l’RNA antisenso continua ad essere espresso e ad indurre sintesi di distrofina in tutti i distretti muscolari inclusi il cuore e il diaframma. La prolungata espressione di distrofina e i livelli di proteina prodotti sono inoltre tali da preservare questi muscoli dalla forte degenerazione tipica dei topi mdx della stessa eta’ e da migliorarne le capacità funzionali.
Altri tipi di esperimenti di terapia genica hanno dimostrato che, nelle scimmie, i geni veicolati mediante AAV rimangono stabili e attivi per oltre 6 anni.
La strategia di exon skipping mediante l’utilizzo di AAV è quindi assai promettente ai fini di una possibile terapia genica della DMD nell’uomo. L’aspetto più entusiasmante di questa tecnica è proprio l’efficacia a lungo termine. A differenza degli approcci terapeutici di exon skipping basati sugli AON, per i quali la somministrazione delle molecole antisenso avviene quotidianamente per tutto il periodo di trattamento, la strategia italiana lascia sperare per una possibile terapia basata su di un trattamento che viene ripetuto solo ogni 5-6 anni.
Adesso, il percorso per accedere alla sperimentazione sull’uomo prevede ulteriori fasi di sperimentazione pre-clinica per ottimizzare l’efficacia dei vettori virali e delle molecole da utilizzare per ottenere lo skipping di diversi esoni della distrofina, e per verificare la tossicità di questo trattamento. In particolare, il gruppo italiano sta ora testando diverse molecole di RNA antisenso per lo skipping dell’esone 51 e analizzando la possibilita’ di ideare nuove molecole per lo skipping di esoni diversi, l’obiettivo finale e’ di progettare una terapia che possa essere utilizzata per diversi profili genetici e quindi per un numero maggiore di pazienti DMD.
5- L'exon skipping francese
 Olivier Danos
Olivier Danos Alla sana competizione “il miglior exon skipping” partecipa anche un gruppo di ricerca francese, che da anni utilizza vettori AAV per veicolare all’interno delle cellule muscolari geni terapeutici. Olivier Danos, dell'Inserm di Parigi, ha mostrato come esperimenti preliminari dimostrano che l’iniezione nella vena femorale di AAV veicolante il gene U7snRNA porta alla produzione di una forma corta, ma funzionale, della distrofina in gran parte dei muscoli dell’arto trattato. Purtroppo l’approccio mediante AAV pone il problema della risposta immunitaria, un ostacolo che subentra nel caso di iniezioni ripetute del vettore. Il virus ha un involucro proteico che può scatenare una difesa da parte dell’organismo che lo riceve. Per la prima somministrazione non dovrebbero esserci problemi, il sistema immunitario infatti non è ancora fornito degli anticorpi necessari, ma per le somministrazioni successive - quando l’organismo e’ oramai istruito a reagire contro il corpo virale estraneo – necessitano dei trattamenti di immuno-soppressione del paziente.
Benché per gli AAV la risposta immunitaria sia inferiore rispetto a quella generata da altri vettori virali usati in passato, il problema va comunque affrontato e risolto. Su questo si e’ basato l’ultimo lavoro di Olivier Danos, del gruppo di ricerca di Luis Garcia, pubblicato a gennaio su Molecular Therapy. Lo studio, effettuato su topi mdx, ha valutato la possibilità di inoculare il vettore AAV1 mediante ripetute iniezioni intramuscolari. I dati hanno dimostrato che l’introduzione del vettore virale nell’organismo scatena una risposta immunitaria che preclude la possibilità di ri-somministrare il vettore oltre 3 giorni dopo la prima iniezione. Quello che è stato scoperto dai ricercatori è che questa risposta dipende unicamente dalla produzione di anticorpi specifici contro AAV1.
Strategie messe a punto per bloccare i segnali cellulari che stimolano il riconoscimento degli anticorpi virali e l’innesco di una risposta immunitaria hanno dato dei risultati molto incoraggianti. Questi esperimenti rappresentano un punto di partenza per la messa a punto di protocolli di immuno-modulazione, ovvero di sistemi che tengano a bada il sistema immunitario, per permettere una sicura applicazione della terapia genica basata su AAV in pazienti DMD.
di Francesca Ceradini
DIAGNOSI GENETICA PRE-CONCEPIMENTO
 Lo scorso 29 febbraio, per via di una conferenza stampa, è stata data la notizia di una nuova procedura che consente di diagnosticare tutti i tipi di malattie genetiche e cromosomiche a trasmissione materna senza toccare gli embrioni. La tecnica, chiamata “diagnosi pre-concepimento” e pubblicata sul numero di gennaio della rivista scientifica Prenatal Diagnosis, è stata messa a punto da un team italiano coordinato da Francesco Fiorentino, biologo molecolare e direttore del Laboratorio Genoma di Roma.
Lo scorso 29 febbraio, per via di una conferenza stampa, è stata data la notizia di una nuova procedura che consente di diagnosticare tutti i tipi di malattie genetiche e cromosomiche a trasmissione materna senza toccare gli embrioni. La tecnica, chiamata “diagnosi pre-concepimento” e pubblicata sul numero di gennaio della rivista scientifica Prenatal Diagnosis, è stata messa a punto da un team italiano coordinato da Francesco Fiorentino, biologo molecolare e direttore del Laboratorio Genoma di Roma.
In Italia la legge 40 sulla fecondazione assistita è molto controversa e, fino ad oggi, ha vietato la diagnosi pre-impianto. La problematica etica riguarda la possibilità di selezionare gli embrioni con l'eliminazione di quelli malati.
La diagnosi genetica pre-concepimento viene eseguita sull’ovocita e non sull’embrione. In particolare, la tecnica permette di studiare i gameti femminili prima della loro fertilizzazione in vitro con lo scopo di escludere quegli oociti il cui DNA risulta alterato alla diagnosi, ed evitare quindi a priori la possibilità di produrre embrioni con anomalie genetiche. In quest’ottica la diagnosi pre-concepimento consente di superare gli ostacoli etici.
“Con questo metodo siamo riusciti ad ottenere una prima gravidanza in una donna laziale portatrice per la sindrome Charcot Marie Tooth. Reduce da gravidanze andate male e da viaggi all’estero, la donna è oggi al suo terzo mese e i dati ottenuto dalla villocentesi hanno confermato che il feto è sano”, ha dichiarato Francesco Fiorentino.
La procedura, che è stata finora effettuata in via sperimentale, è stata validata a livello internazionale ed è ora in fase di proposta per l’attuazione in ambito sanitario. Le patologie genetiche per le quali la diagnosi pre-impianto potrebbe trovare una valida applicazione comprendono quelle a trasmissione autosomica recessiva, come ad esempio la fibrosi cistica, la talassemia e la SMA, o quelle di origine materna, quali la distrofia muscolare di Duchenne-Becker e l’X-Fragile.
Secondo Francesco Fiorentino questa nuova tecnica ha le potenzialità di dare, in un futuro prossimo, una reale possibilità alle coppie italiane portatrici di queste patologie di avere figli sani, senza dover ricorrere a costosi viaggi all'estero per ottenere trattamenti vietati in Italia.
Francesca Ceradini
Intervista a Francesco Fiorentino su RadioRadicale
Bibliografia
Prenatal Diagnosis - 2008 Jan;28(1):62-4
Rapid protocol for pre-conception genetic diagnosis of single genemutations by first polar body analysis: a possible solution for the Italian patients
F. Fiorentino 1 2, A. Biricik 1, A. Nuccitelli 2, R. De Palma 2, S. Kahraman 3, S. Sertyel 3, H. Karadayi 3, G. Cottone 2, M. Baldi 1 2, D. Caserta 4, M. Moscarini 4
1 EmbryoGen - Centre for Preimplantation Genetic Diagnosis, Via Po, 102 00198 Rome, Italy
2 GENOMA- Molecular Genetics Laboratory, Via Po, 102 00198 Rome, Italy
3 ART and Reproductive Genetics Unit, Istanbul Memorial Hospital, Istanbul, Turkey
4 University of Rome La Sapienza - Department of Gynaecological Science, Perinatology and Child Care, S. Andrea Hospital, via di Grottarossa, 1035 00189 Rome, Italy
