

ReveraGen BioPharma riceve 3M di dollari per lo studio di fase 2 con il Vamorolone nella Duchenne
Il National Institute of Neurological Disorders and Stroke (NINDS) del National Institut of Health ha assegnato un finanziamento da 3M di dollari a ReveraGen BioPharma per lo svolgimento di studi clinici con il vamorolone (precedentemente VBP 15) nei ragazzi con distrofia muscolare di Duchenne (DMD). La DMD è causata dall'assenza della proteina distrofina che determina un progressivo danno muscolare, debolezza e disabilità. Il Vamorolone rallenta questo danno muscolare nei modelli murini della DMD.
Questi studi con il vamorolone, per la prima volta nei pazienti, seguono il completameno positivo dello studio di fase 1 in volontari adulti che ha mostrato la sicurezza e la tollerabilità del vamorolone. Studi di fase 2a, con dosaggi multipli crescenti (MAD - multiple ascending dose), saranno condotti in ragazzi Duchenne con età tra i 4 e <7 anni, a questi studi seguirà uno studio di estensione della durata di 6 mesi. Nelle prime due settimane della fase MAD saranno studiate la sicurezza, tollerabilità e farmacocinetica. L'efficacia clinica, la sicurezza e biomarcatori farmacodinamici saranno valutati nei 6 mesi dello studio di estensione, con l'obiettivo di selezionare il dosaggio per i futuri studi clinici.
Il Vamorolone è il primo nell'uomo agente steroideo dissociativo che mira a separare le problematiche relative alla sicurezza dei farmaci glucocorticoidi tradizionali come il prednisone e il deflazacort, dagli aspetti chiave di efficacia. Negli studi pre-clinici nei modelli murini della distrofia muscolare e di patologie infiammatorie il vamorolone ha mantenuto l'efficacia perdendo allo stesso tempo gli effetti collaterali come l'arresto della crescita.
Eric Hoffman CEO di ReveraGen, e sperimentare principale del finanziamento NINDS ha commentato "I dati della fase 1 supportano i nostri risultati precedenti nei modelli pre-clinici, tra i quali l'assenza di risultati che suggeriscano l'insulino-resistenza e una ridotta soppressione dell'asse surrenale. Lo studio clinico di fase 2a inizierà ad aiutare la validazione del profilo migliore di sicurezza nei ragazzi DMD e a fornire conoscenze circa la dose appropriata per gli studi di efficacia".
John McCall, chimico principale e co-fondatore del programma vamorolone ha detto "Siamo grati per l'assistenza dell'NIH nel far avanzare il programma clinico. Questo nuovo finanziamento dell'NINDS segue collaborazioni chiave con il programma NCATS TRND dell'NIH e il supporto economico fondamentale di organizzazioni no-profit negli USA, UK e Australia".
Il nuovo finanziamento comprende ampi studi di farmacodinamica, sicurezza e biomarcatori di efficacia. Kanneboyina Nagaraju co-fondatore di ReveraGen ha detto "la nostra inclusione dei biomarcatori nel programma con il vamorolone promette di fornire risultati acuti e obiettivi sull'attività della molecola". Lo studio clinico di fase 1 è stato supportato dalla statunitense Muscular Dystrophy Association e da tre fondazioni inglesi (Joining Jack, Duchenne Children's Trust e Duchenne Research Fund). La Dott. Paula Clemens della University of Pittsburgh condurrà lo studio di fase 2a che coinvolgerà ragazzi DMD presso 8 centri negli USA del Cooperative International Neuromuscular Research Group (CINRG). La Dott. Clemens ha osservato "gli studi di fase 2 che stanno per iniziare sono stati pianificati accuratamente nel corso dell’ultimo anno insieme al NIH National Institute of Arthrites and Muscoskeletal ( NIAMS) che ha fornito un finanziamento critico attraverso un fondo per la pianificazione di studi clinici per supportare il progetto " La Dott. Clemens è anche sperimentatore principale nel finanziamento NINDS dello studio clinico. Il reclutamento iniziale dei ragazzi DMD con età tra i 4 e <7 anni è previsto nel secondo trimestre del 2016.
Traduzione a cura dell’Ufficio Scientifico di Parent Project Onlus

Notiziario #1 2016
http://issuu.com/parentprojectonlus/docs/notiziario1_2016-translarna-2?workerAddress=ec2-54-210-46-23.compute-1.amazonaws.com
Summit annuncia i dati intermedi positivi dello studio clinico di fase 1 in corso dedicato alla valutazione di una nuova formulazione di SMT C1100 nei pazienti DMD.
Oxford, Regno Unito 30 marzo 2016 -Summit Therapeutics plc (NASDAQ: SMMT, AIM: SUMM), la company dedicata alla scoperta e sviluppo di farmaci per l'avanzamento delle terapie per la distrofia muscolare di Duchenne e l'infezione da Clostridium difficile, annuncia i dati positivi intermedi provenienti dallo studio clinico di fase 1 che sta valutando una nuova formulazione orale del suo principale modulatore dell'utrofina, SMT C1100 per la DMD. In questo periodo iniziale di somministrazione dello studio clinico con aumento graduale della dose, tutti i pazienti hanno raggiunto livelli della molecola nel plasma entro l'intervallo ritenuto necessario per un potenziale beneficio terapeutico. La dose iniziale valutata della nuova formulazione è stata un decimo di quella necessaria con la formulazione attuale di SMT C1100 per raggiungere livelli di concentrazione simili a quelli dello studio clinico recente di fase 1b con dieta modificata di SMT C1100.
SMT C1100 è una potenziale terapia per modificare la patologia per tutti i pazienti DMD e sta avanzando con la formulazione attuale, verso uno studio clinico di fase 2 finalizzato a confermare il meccanismo di azione.
Ralf Rosskamp, MD, Chief Medical Officer di Summit ha commentato “Questi dati iniziali sono incoraggianti rispetto al potenziale sviluppo di una formulazione migliore che potrebbe raggiungere livelli nel sangue di SMT C1100 che sono superiori a quelli osservati con la nostra formulazione attuale. In un precedente studio clinico di fase 1 abbiamo visto come una dieta bilanciata, consenta alla nostra attuale formulazione di raggiungere livelli nel sangue che crediamo potrebbe fornire un beneficio terapeutico e il nostro lavoro in corso di sviluppo della nuova formulazione potrebbe fornire maggiori conoscenze circa i potenziali benefici di SMT C1100 sulla modulazione dell'utrofina nei pazienti DMD".
Lo studio clinico di fase1 con la nuova formulazione di SMT C1100 sta procedendo per valutare un dosaggio più elevato della nuova formulazione. Decisioni definitive rispetto all'ulteriore sviluppo di questa nuova formulazione verranno prese quando saranno disponibili tutti i dati del trial.
Lo studio clinico di fase 1 con la nuova formulazione di SMT C1100
Lo studio clinico di fase 1 con la nuova formulazione di SMT C1100 è costituito da due parti. La parte A ha valutato due nuove formulazioni orali di SMT C1100 in 16 maschi volontari sani. Una di queste nuove formulazioni, che ha raggiunto un aumento superiore alle 10 volte dell’esposizione nel plasma dei volontari sani rispetto alla formulazione attuale di SMT C1100, è stata selezionata per progredire nella parte B dello studio.
La parte B del trial sta valutando la farmacocinetica e la sicurezza di fino a tre livelli di dosaggio di una nuova formulazione di SMT C1100 presa due volte al giorno per sette giorni in otto ragazzi DMD di età tra i cinque e i nove anni. Nel periodo iniziale della somministrazione della nuova formulazione nel trial di fase 1, i pazienti che hanno ricevuto un dosaggio fisso di 250mg della nuova formulazione di SMT C1100 per sette giorni hanno raggiunto livelli plasmatici della molecola simili a quelli dei pazienti che hanno ricevuto un dosaggio fisso di 2,500mg della formulazione attuale di SMT C1100 nello studio di fase 1b con dieta modificata completato nel 2015. La nuova formulazione di SMT C1100 è stata generalmente ben tollerata nei volontari sani e nei pazienti che hanno ricevuto il primo livello di dosaggio della nuova formulazione di SMT C1100. I pazienti in questo trial di fase 1 stanno seguendo indicazioni dietetiche simili a quelle del precedente studio di fase 1b con dieta modificata.
Traduzione a cura dell’Ufficio Scientifico di Parent Project Onlus
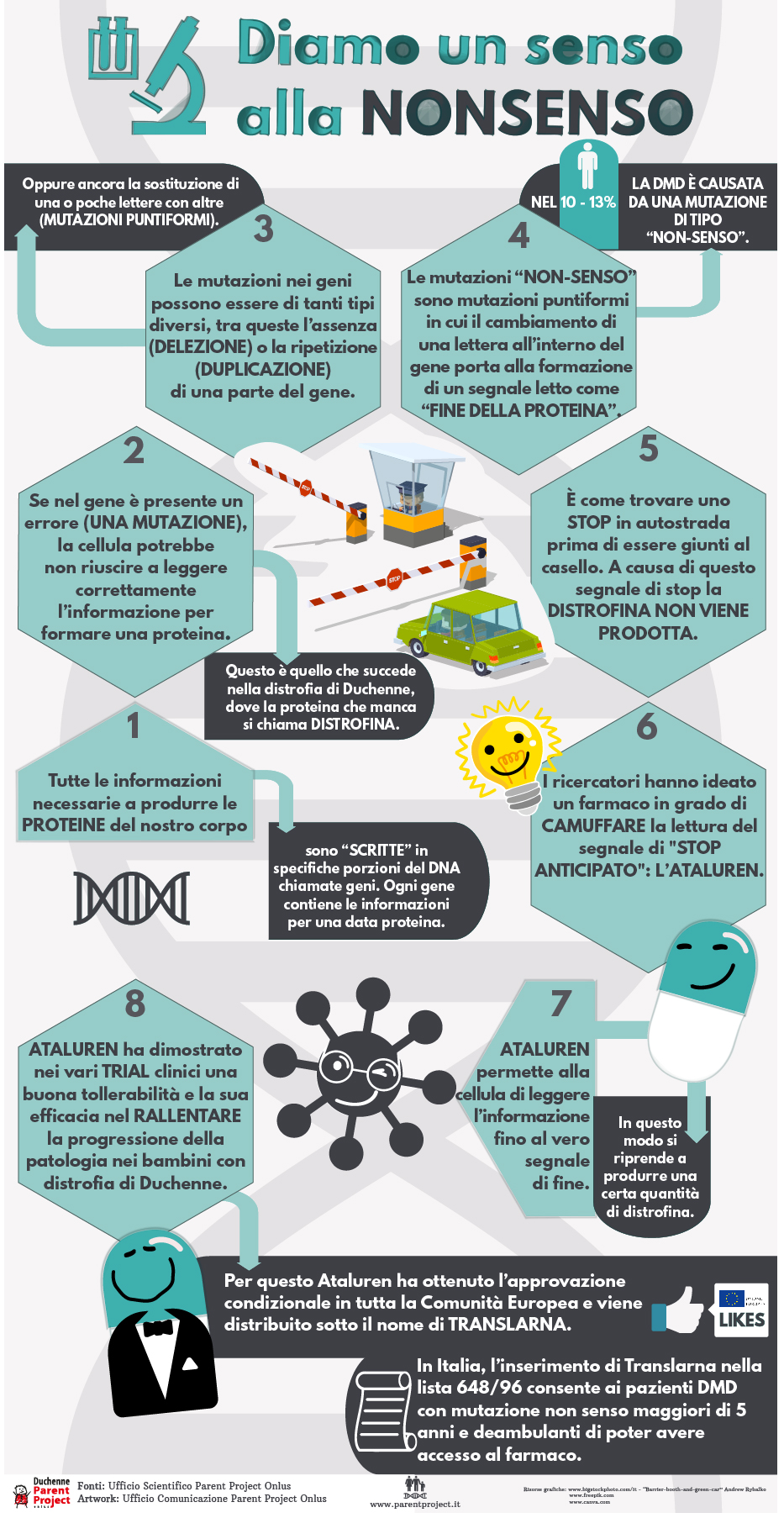
Diamo un senso alla nonsenso
La formazione è parte integrante delle attività di Parent Project. E’ per questo motivo che diamo il via ad un programma formativo, su specifici argomenti, orientato sull’utilizzo di uno strumento semplice ed innovativo: l’infografica.
L'infografica (anche nota con termini inglesi "information design", information graphic o infographic) è l'informazione proiettata in forma più grafica e visuale che testuale. Come tecnica è nata dall'incrocio delle arti grafiche con il giornalismo e l'informatica.
Le immagini insieme a frasi brevi ed efficaci, ci guideranno nell’approfondimento di concetti legati alla distrofia muscolare di Duchenne e Becker.
Apriamo il programma parlando di mutazione nonsenso : diamo un senso alla nonsenso!
Buona lettura!
FORTI, VELOCI E… SOLIDALI: UNA SQUADRA DI RUNNER AL FIANCO DI PARENT PROJECT
Lo scorso 13 marzo, un gruppo di atleti molto speciale ha corso alla RomaOstia, la celebre mezza maratona che registra ogni anno un record in termini di partecipazione: è il team di “Valore Salute Forti e Veloci”, associazione sportiva che ha scelto di sostenere, attraverso Parent Project onlus, la ricerca sulla distrofia muscolare di Duchenne e Becker.
La squadra ha donato all’associazione il costo del pettorale e ha organizzato, la sera del 12 marzo, una grande serata di solidarietà a supporto della ricerca sulla patologia.
“La nostra storia è iniziata nel 2009” racconta il Presidente, Carlo Sensi “quando Valore Salute Forti e Veloci è prima nata come un progetto ambizioso, e poi è stata costituita come associazione per la promozione di uno stile di vita sano incentrato sulla prevenzione.
Il 2009 era l’anno della sindrome Metabolica, che si può combattere solo cambiando diametralmente stile di vita, grazie all’attività fisica e ad un’alimentazione sana. Noi ci siamo impegnati in prima linea sul tema, come Network dedicato alla salute e al benessere, nell’organizzazione dei corsi ECM rivolti ai Farmacisti. Abbiamo realizzato in parallelo un’importante campagna di informazione al cittadino, denominata Togli centimetri, Aggiungi salute, veicolata sui media nazionali. Proprio mentre eravamo impegnati alla creazione di questa campagna per la salute, ci venne un’idea che in realtà era una sfida: per essere d’esempio noi per primi, dovevamo passare ai fatti, iniziare ad allenarci, prepararci e partecipare alla maratona di New York, il più importante e significativo evento mondiale per i podisti. Il nostro obiettivo era cambiare stile di vita noi in prima persona e “Allenarci a stare bene!”. Dieci mesi dopo, con un bel bagaglio di fatica, sudore, chilometri, regole, chili di meno, consigli e grande entusiasmo, siamo atterrati nella Grande Mela e tra lo stupore di tutti, noi per primi, abbiamo corso e tagliato il traguardo della maratona più importante del mondo. L’energia e la carica erano a mille e hanno fatto nascere una nuova ambiziosa idea: abbiamo deciso di costituire un’associazione sportiva per promuovere la corsa come elemento fondamentale di salute e strumento di prevenzione e benessere psicofisico. Il 21 marzo 2010 abbiamo partecipato alla Maratona di Roma migliorando ancora i nostri tempi e abbiamo presentato ufficialmente la neonata associazione: Valore Salute Forti e Veloci. Da allora abbiamo partecipato a numerose gare e maratone”.
L’incontro con Parent Project avviene attraverso il contatto con Filippo Buccella: “Mostrandomi il filmato di suo figlio Luca, Filippo mi ha coinvolto nel suo grande impegno per Parent Project. Tutto questo non mi ha lasciato indifferente e mi ha aiutato a riflettere su questa tema. La capacità di muoversi e di fare esercizio aiuta la nostra forma fisica, ed è in questo modo che pensare alla DMD diventa proprio complementare: ho dedotto che la nostra forza muscolare potesse offrire un aiuto in più ai bambini affetti da questa malattia”.
A livello personale, una gara collegata ad una causa, ad un’iniziativa di raccolta fondi viene vissuta in modo diverso dall’atleta, racconta Carlo Sensi: “Si trova più forza nei propri muscoli e si esprimono al massimo le proprie capacità dilettantistiche, sentendosi più utili. Questa gara è stata affrontata con un grande spirito di gruppo, nonostante il running sia uno sport tendenzialmente solitario. L’evento è stato vissuto in questa maniera: abbiamo corso per il gruppo e non per noi stessi. In futuro ci piacerebbe dedicare ogni edizione della Roma Ostia alla ricerca scientifica. ‘Forti nel cuore e veloci nelle gambe’: è questa la nostra ambizione. Basta un piccolo sforzo per regalare un sorriso e noi vogliamo dirlo a voce piena”.
Un grosso grazie da parte dell’associazione per il sostegno ricevuto finora e…arrivederci alla prossima impresa sportiva!
Biomarker per la DMD: la ricerca continua
Sono sempre di più i nuovi biomarker candidati per la distrofia muscolare di Duchenne e Becker, ma al momento nessuno è ancora stato validato come strumento diagnostico e di monitoraggio della malattia. Tre recenti studi internazionali puntano l’attenzione verso diverse proteine che sembrano correlare molto bene con le diverse fasi della distrofia.
Francesca Ceradini
Con l’avanzamento della ricerca scientifica e il costante incremento di nuovi trial clinici in avvio per la DMD e BMD, individuare e validare nuovi biomarker specifici per la patologia sta diventando un’esigenza sempre più urgente. Trovare i biomarker giusti vorrebbe dire poter diagnosticare e seguire l’andamento della malattia in maniera molto accurata, e soprattutto poter valutare e validare gli effetti terapeutici di nuovi trattamenti in sperimentazione.
Ma facciamo un attimo un salto indietro e partiamo dalle basi: cosa sono effettivamente i biomarker? In italiano vengono anche comunemente chiamati biomarcatori o marcatori biologici e sono quelle molecole, normalmente presenti nel nostro organismo, che possono essere misurate e monitorate per fornire informazioni sui processi patologici, come ad esempio determinare il tipo di malattia e lo stadio di progressione. I biomarker possono essere proteine, o acidi nucleici come l’RNA o DNA, presenti nei liquidi corporei (sangue, urine, saliva) così come nei tessuti o nelle cellule. La distrofina è il biomarker per eccellenza per la distrofia muscolare di Duchenne e Becker: la totale assenza di questa proteina nelle cellule muscolari indica la DMD, mentre una parziale presenza indica la BMD, forma meno grave della malattia. La diagnosi clinica della Duchenne e Becker mediante la quantificazione della distrofina comporta però una biopsia muscolare, e lo stesso spesso avviene per la valutazione di una nuova strategia terapeutica durante un trial. Un bambino che partecipa a uno studio clinico potrebbe, ad esempio, avere fatto in tutto tre biopsie muscolari, una per la diagnosi e due nel corso del trial clinico. Un approccio un po’ troppo invasivo, che pone dei problemi anche etici e per cui la comunità scientifica sta cercando di trovare una valida alternativa: nuovi biomarcatori affidabili, facilmente misurabili, presenti nel sangue, urine o saliva dei pazienti.
A questo riguardo negli ultimi anni sono stati condotti molti studi, alcuni anche a livello di network europei come ad esempio il progetto BIO-NMD finanziato dal Settimo Programma Quadro, e sono state individuate diverse molecole promettenti (tra cui alcuni microRNA) ma nessuna è ancora stata validata ufficialmente come strumento diagnostico e di monitoraggio della malattia. Il lavoro va quindi avanti e i ricercatori continuano a individuare e valutare nuovi biomarker. Recentemente sono stati pubblicati tre studi che propongono nuovi candidati, si tratta di proteine che sono presenti nel sangue e la cui quantità correla bene con la progressione della malattia. Un primo lavoro, che nasce dalla collaborazione tra l’Università di Newscastle e l’azienda farmaceutica Pfizer, è stato pubblicato su “Journal of Neuromuscular Diseases” e ha monitorato la presenza nel sangue di quattro proteine, normalmente prodotte nei muscoli, in pazienti con tre diverse distrofie muscolari. Un secondo studio, pubblicato su “skeletal Muscle”, è stato condotto da gruppi di ricerca brasiliani e francesi e ha invece puntato l’attenzione su una proteina che partecipa alla risposta immunitaria dei linfociti T. L’ultimo è un lavoro giapponese pubblicato su “The American Journal of Pathology” e propone come biomarker una citochina coinvolta nella rigenerazione muscolare.
Nel primo lavoro i ricercatori hanno condotto lo studio su 161 pazienti: 74 con la DMD, 38 con la BMD e 49 con la distrofia muscolare dei cingoli (LGMD), e su 38 volontari sani. Hanno monitorato quattro diverse proteine: la troponina I (TnI), che ha una funzione nella contrazione muscolare, la catena leggera 3 della miosina (Myl3), importante per la formazione del complesso contrattile del muscolo, la proteina legante gli acidi grassi (FABP3), che ha un ruolo nel trasporto degli acidi grassi attraverso la membrana cellulare, e la creatinchinasi specifica del muscolo (CKM), un enzima coinvolto nel meccanismo energetico della cellula. Il primo dato saliente è che tutte e quattro le proteine sono presenti nel sangue in concentrazioni molto più elevate nei pazienti rispetto ai soggetti sani e, in particolar modo, la quantità riflette la gravità e quindi il tipo di distrofia muscolare. Ad esempio, per CKM la concentrazione nel sangue dei pazienti DMD è ben 88 volte superiore rispetto alle persone sane, nei pazienti LGMD parliamo di un fattore 37 e nei pazienti BMD di un fattore 17. I ricercatori sono andati oltre a questi dati, importanti ma solo indicativi, e sono riusciti a determinare una stretta correlazione tra la quantità di questi biomarker nel sangue e la fase di progressione della malattia, soprattutto nel caso della DMD. Una prima osservazione è stata fatta riguardo alla deambulazione: i pazienti DMD ancora in grado di camminare, e quindi nella fase iniziale della malattia, presentano quantità più elevate di tutte e quattro le proteine rispetto ai ragazzi che hanno perso la deambulazione. Un dato che rispecchia nozioni già note, ovvero che con l’esercizio fisico e la contrazione muscolare si ha un maggiore rilascio di proteine dei muscoli nel sangue. Con la perdita di deambulazione e il progredire della Duchenne i pazienti vanno solitamente incontro a una diminuzione della massa muscolare, un indebolimento dei muscoli respiratori e l’insorgenza di cardiomiopatia. I ricercatori sono riusciti a dimostrare un legame tra tutti questi fattori e la quantità rilevabile dei biomarker nel sangue. Nello specifico, con l’aumento dell’età del paziente e la comparsa di sintomi strettamente legati alla degenerazione del tessuto muscolare si ha una progressiva diminuzione dei biomarker. Nessuna correlazione è stata invece dimostrata tra il trattamento con steroidi e la variazione delle concentrazioni delle proteine in questione. Gli autori del lavoro suggeriscono che con studi più approfonditi si potrebbe riuscire a determinare dei valori soglia di questi candidati biomarker che potrebbereo essere associati a specifiche distrofie e al loro grado di progressione.
A differenza di questo studio, quello portato avanti dai ricercatori brasiliani e francesi è focalizzato su un’unica proteina: CD49d. Si tratta di una componente delle integrine, le proteine responsabili dei meccanismi di interazione cellula-cellula o cellula-matrice extracellulare (ovvero l’ambiente circostante). Tra le varie funzioni, questi meccanismi di interazione sono anche alla base di molti processi cellulari tra i quali la risposta immunitaria, l’infiammazione e la rigenerazione di un tessuto. Il punto di partenza del lavoro pubblicato è l’osservazione che nei pazienti Duchenne si ha un’alta circolazione nel sangue di alcune cellule immunitarie, nello specifico di linfociti T CD4 e CD8 alle quali è legata la proteina CD49d. I ricercatori hanno approfondito il dato analizzando le concentrazioni di CD49d in 75 pazienti DMD, suddivisi in tre gruppi in base alla progressione della malattia (non deambulanti, in grado di percorrere 10m in meno di 10 secondi, in grado di percorrere 10m in più di 10 secondi). I risultati dimostrano che la quantità di CD49d nel sangue è correlata con lo stadio della malattia. I ragazzi non deambulanti hanno concentrazioni della proteina ben maggiore di quelli deambulanti, e tra questi la concentrazione aumenta con l’aumentare delle difficoltà nel camminare. Ma l’osservazione non si è fermata qui, i ricercatori hanno seguito negli anni 23 bambini DMD, dalla fase in cui ancora camminano alla fase in cui devono ricorrere alla carrozzina. L’andamento di espressione di CD49d nel sangue riflette perfettamente la progressione della Duchenne, un dato saliente per un futuro utilizzo di questa proteina nella valutazione di nuove terapie in sperimentazione. Anche in questo caso, gli autori non hanno però evidenziato nessuna correlazione tra l’assunzione di steroidi come trattamento per la patologia e la variazione delle concentrazioni di CD49d. Basandosi su precedenti osservazioni della presenza di linfociti T esprimenti CD49d all’interno degli infiltrati infiammatori nei muscoli, i ricercatori hanno ipotizzato un ruolo di questa proteina nel processo di infiammazione muscolare tipico della patologia. Con una serie di esperimenti hanno infatti dimostrato che CD49d interagisce con la matrice extracellulare e accelera il meccanismo di migrazione delle cellule immunitarie, alle quali è legata la proteina, verso le fibre muscolari potenziando il processo infiammatorio. Bloccando l’azione di CD49d la capacità di migrazione e il grado di infiammazione diminuiscono. Lo studio suggerisce quindi che CD49d, oltre ad essere un ottimo candidato biomarker, potrebbe anche rivelarsi un bersaglio per una nuova strategia terapeutica basata sulla riduzione della risposta immunitaria e del conseguente processo infiammatorio. Strategia che se abbinata ad altri approcci, più mirati al difetto genetico (exon skipping, terapia genica), potrebbe risultare vincente.
Anche il terzo lavoro ha puntato l’attenzione su una proteina, l’osteopontina (OPN), che si lega alle cellule immunitarie (CD44) e che è coinvolta nei processi infiammatori via meccanismi di interazione cellula-cellula o cellula-matrice extracellulare. Questa volta però lo studio non è stato condotto su pazienti ma su cani distrofici. I ricercatori hanno osservato che le concentrazioni di osteopontina rilevate nel sangue di cani distrofici, appena nati e fino a 3 mesi di età, sono nettamente superiori a quelle trovate in cani sani nella stessa fase di sviluppo. Inoltre, i livelli di osteopontina correlano bene con il grado di severità della distrofia. Studi precedenti hanno ipotizzato che l’osteopontina non agisca solo nel processo di infiammazione muscolare ma anche in quello rigenerativo, dato che è stato confermato da esperimenti condotti dai ricercatori giapponesi. Nello specifico, gli autori hanno utilizzato la cardiotossina, una sostanza contenuta nel veleno del cobra, per indurre un danno nel tessuto muscolare di cani sani e hanno successivamente osservato non solo la presenza di osteopontina nelle cellule immunitarie all’interno degli infiltrati infiammatori nei muscoli ma anche un notevole incremento di espressione nelle fibre in rigenerazione e nel sangue circolante. Nonostante questi risultati vengano da uno studio su modelli animali, e quindi un passo indietro rispetto agli altri due studi descritti su pazienti, la loro valenza risiede nel fatto che osservazioni simili, anche se meno dettagliate, erano state fatte qualche anno fa da un altro gruppo di ricerca su pazienti Duchenne. I giapponesi hanno infine dimostrato che c’è una stretta correlazione tra alti livelli di osteopontina nel sangue e la fase di rigenerazione muscolare associata alla DMD, lo stadio iniziale della malattia, ma non con la fase tardiva cronica, ovvero quando la rigenerazione muscolare è ormai inattiva. Su questa base il lavoro suggerisce che l’osteopontina potrebbe essere un utilissimo biomarker per valutare strategie terapeutiche che puntano ad agire sulla rigenerazione muscolare.
Un ultimo importante aspetto riguardo a questi tre lavori, presi nel loro insieme, è che tutte le proteine studiate sono conservate funzionalmente, ovvero hanno lo stesso ruolo, dai modelli animali utilizzati per la Duchenne, quali i topi e i cani, agli umani. Sono quindi degli ottimi candidati biomarker perché possono essere utilizzati trasversalmente nel percorso della ricerca traslazionale, ovvero dalla ricerca preclinica fino agli studi clinici. Un aspetto fondamentale per la loro validazione e successiva qualifica a livello internazionale da parte delle agenzie regolatorie (FDA, EMA) come strumento diagnostico e di monitoraggio della malattia. La speranza è che, in un futuro prossimo, si potrà monitorare la progressione della patologia in modo accurato permettendo una migliore gestione dei sintomi clinici e, perché no, permettendo di personalizzare una terapia (tipo e dosaggio del farmaco) a seconda della risposta del paziente ai trattamenti esistenti.
Un nuovo presidente per Parent Project Onlus
Cari amici, associati, volontari e staff,
Innanzitutto penso sia giusto presentarmi; non ho certo la fama di Filippo!! Mi chiamo Luca Genovese, ho 51 anni, dei quali almeno 15 trascorsi a confrontarmi quotidianamente con la “Duchenne”. Questo strano termine, entrato brutalmente nelle nostre vite ma che ha via via assunto, nel corso di questi 15 anni, sfumature diverse. Dal “nero” notte fonda post diagnosi, al “grigio” degli anni della presa di coscienza e della reazione, all’“avorio” della fase della stabilizzazione, fino al “rosa” color pelle. Si, perché oggi sento che la Duchenne mi è proprio “entrata nella pelle”. Fa parte della mia, delle nostre esistenze e con essa si convive, magari a volte non “allegramente”, ma certo “serenamente”.
In questo processo, lungo 15 anni, di sicuro Parent Project ha rappresentato per me un punto di riferimento importante, una fonte sicura di informazioni corrette, il giusto pungolo per un mondo della ricerca spesso “stagnante” (di certo lo era 15 anni fa; oggi, per fortuna, è in grande ebollizione!), l’importante collante tra ricercatori e clinici. Parent Project Onlus ha anche rappresentato per tutti noi l’occasione per il confronto e per l’individuazione delle migliori pratiche per la gestione dei nostri figli.
Si…., perché io di ragazzi Duchenne ne ho due, Matteo, 21 anni e Marco, 17.
Inutile dire quanto mi senta in debito con Filippo Buccella per quanto fatto in tutti questi anni. Inizialmente coadiuvato dalla “storica” Silvia Starita e da un manipolo di “genitori coraggiosi e forti” che hanno operato nelle proprie famiglie e nei propri territori. Se oggi Parent Project Onlus è quella che è, una grande e composita realtà associativa, è proprio grazie a queste persone, “grandi” nella loro normalità, “normali” nella loro grandezza.
E’ in questo contesto e con un’adeguata dose di gratitudine (accompagnata forse da altrettanta dose di incoscienza, ma questo lo lascio “tra parentesi”) che, comprendendo le ragioni di Filippo – da 20 anni in trincea con l’elmetto – ho accettato di dare una mano per come oggi mi viene chiesto: assumere la presidenza di Parent Project Onlus per un periodo definito. Un periodo durante il quale l’associazione deve continuare il proprio percorso di crescita per diventare una struttura matura ed organizzata, capace di andare oltre le persone!!! Perché ovviamente il sogno sarebbe che Parent Project si sciogliesse domattina per “avvenuto conseguimento dell’oggetto sociale”, ovvero un mondo libero dalla distrofia di Duchenne e di Becker, ma purtroppo realisticamente sappiamo che questo traguardo, oggi seriamente ipotizzabile, non potrà essere raggiunto in breve tempo. Quindi l‘associazione deve proiettarsi nell’ottica di media-lunga durata e questo significa che non si potrà contare sempre sulle stesse persone, ma in una sorta di passaggio di testimone tra genitori “presenti”.
Non sarebbe giusto però nascondere che nonostante gli oneri, assumere la carica di Presidente di Parent Project sia per me anche e certamente un onore. Parent Project Onlus oggi è un’associazione autorevole nel proprio campo, di portata nazionale ed internazionale e con collegamenti in tutto il mondo. Ma proprio per questo mi auguro di essere all’altezza del ruolo, perché la responsabilità che avverto è grande, ma per fortuna le motivazioni interiori lo sono di più. Ed in questo mi dà conforto la presenza di una struttura già ben organizzata e ben coordinata. Composta da gente che vive la problematica con forza e motivazioni pari a quelle di genitori.
Inoltre mi dà conforto la certezza che il buon Filippo non tirerà affatto i remi in barca, anzi penso proprio che, libero dalle pastoie burocratiche e formali, possa dispiegare con maggior vigore la sua forza propulsiva in ciò che sta a cuore a tutti noi: la ricerca di una terapia per la Duchenne e la Becker. Mi danno forza anche i colleghi del Consiglio Direttivo (“colpevoli” di avermi individuato per questo passaggio di testimone). Tutti continueranno col loro impegno per “caricarsi” ciascuno un po’ di peso, Stefano Mazzariol in testa, in qualità di vice.
Per completare il quadro della presentazione e salutarvi tutti affettuosamente, debbo anche presentarvi il resto della mia famiglia e non è poco. Mia moglie Franca (che penso non mi consideri del tutto “sano di mente” ad aver accettato questo incarico) e mia figlia Marianna (13 anni), oltre alla nostra fedele Zoe, un golden retriver che da tre anni riempie ulteriormente la nostra casa.
Un abbraccio forte a tutti, un caloroso augurio per una Pasqua all’insegna della serenità e, a tutti, un buon lavoro per …..“fermare la duchenne”.
Luca Genovese
Design for Duchenne: intervista a Michele Marchi
Come è nata la tua collaborazione con Parent Project al progetto dal quale è nato "Design for Duchenne"?
La collaborazione con Parent Project onlus è nata inizialmente in maniera casuale: nel 2011 ho vinto la graduatoria per accedere ad un dottorato in Tecnologia dell’Architettura presso il Dipartimento di Architettura di Ferrara. La ricerca dottorale era cofinanziata tra Parent Project e l’Università di Ferrara. Ricordo con piacere la figura di Fabio Amanti, in quanto era presente nei colloqui finali del concorso. Fin da subito mi sono imbattuto in una realtà a me semi-sconosciuta dal momento che non avevo conoscenze specifiche sulla distrofia di Duchenne e i suoi risvolti psicologici e medici. E’ stata fin da subito una sfida in quanto i “problemi” erano tanti, e gli strumenti informativi o normativi molto deficitari. Nella prima fase di studio della malattia Parent Project e il suo staff sono stati fondamentali.
Che cosa hai provato nel lavorare a questo progetto? Che cosa ti ha dato a livello individuale e come ricercatore?
La notizia di intraprendere una ricerca mirata al soddisfacimento di persone con necessità specifiche ha fatto nascere in me una grande curiosità. Durante gli anni accademici ho perfezionato la conoscenza di un metodo progettuale rivolto al soddisfacimento di quanti più utenti possibili. Tale metodo, chiamato Design for all, mira a progettare e pensare spazi, oggetti e interfacce per un’utenza allargata, senza tralasciare i bisogni più specifici e puntuali. Si è trattato di un percorso formativo che è iniziato durante la mia tesi di laurea con il progetto di un plesso scolastico comprendente asilo nido e scuola dell’infanzia sulle colline forlivesi e che si sta tuttora compiendo con progetti rivolti ad una progettazione ampia, inclusiva ed universale.
Ritengo impensabile, come professionista e come cittadino italiano, che esistano ancora oggi barriere nelle nuove costruzioni. Certamente occorre effettuare un cambio culturale di mentalità e di priorità per cercare di colmare le disuguaglianze presenti, ma il più delle volte tali ostacoli sono di matrice culturale ed informativa. Pensare di progettare spazi e ambienti realmente accessibili per un’utenza complessa, come quella di persone affette da distrofia muscolare di Duchenne, è stato un lavoro stimolante e molto interessante che ha comportato uno sforzo progettuale e culturale importante.
Per cercare di garantire spazi accessibili è stato necessario slegarsi dalla comune prassi progettuale studiata durante i primi anni universitari per cercare nuove distribuzioni e modalità del vivere quotidiano che partissero dalle reali necessità ed aspettative dell’utenza coinvolta.
Il fatto di svolgere una ricerca con un risvolto etico importante è stato determinante, per me, per cercare di non dare nulla per scontato, di alzare l’asticella della qualità del lavoro sempre più in alto, di non accontentarsi mai, di migliorarsi sempre. Auspico che le informazioni trasmesse possano davvero risolvere problemi di natura tecnico e distributiva riguardo le azioni quotidiane delle famiglie Duchenne.
Il tuo lavoro ha prodotto delle linee guida estremamente utili, che potranno trovare applicazione pratica nelle quotidianità di molte persone. Quanto è stato importante per te questo aspetto, nel portare avanti il tuo lavoro su questa tematica, come architetto e come ricercatore?
L’obiettivo della ricerca era quello di produrre strumenti utili e di immediata applicabilità per progettisti e famiglie, che consentano agli uni di interpretare correttamente e consapevolmente il proprio ruolo e alle altre di ottenere servizi, prodotti e spazi adeguati alle proprie esigenze e alle proprie disponibilità economiche.
Data l’importanza della ricerca, per me è stato un obbligo e uno stimolo quello di riuscire a centrare l’obiettivo nei tempi stabiliti.
Svolgere una ricerca con un fine misurabile e concreto è stato sicuramente un fine ambizioso in quanto, a livello internazionale, ad oggi non esistono informazioni simili per una patologia complessa come la DMD e pertanto quella condotta da me è stata una ricerca sperimentale, che partiva da una base di informazioni scarse. La ricerca, inoltre, ha comportato un costante aggiornamento riguardo a nuove scoperte o indicazioni mediche.
Sapere che le informazioni trasmesse sarebbero andate in mano alle famiglie o ai professionisti per modificare i loro spazi domestici mi ha obbligato, da una parte, a fornire informazioni attendibili e di qualità elevata, dall’altra a non dotare il manuale di informazioni prestabilite e standard. Questi due termini non dovrebbero mai essere accomunati nel progetto di ambienti per un’utenza complessa come quella distrofica. Non esistono regole prestabilite per progettare un’abitazione accessibile. E’ sbagliato fornire indicazioni senza conoscere la singola persona con la quale stiamo progettando. Le Linee Guida, pertanto, cercano di informare le famiglie e i professionisti sulle necessità, bisogni ed esigenze presenti e future dei ragazzi DMD e delle loro famiglie; cercano di trasmettere un modus operandi utile per effettuare scelte ragionate in base alle reali necessità.
Il lavoro di ricerca ha premesse molto articolate ma anche molto stimolanti in quanto, oltre alla complessità della malattia, è stato necessario interfacciarsi con una normativa italiana in merito al superamento delle barriere architettoniche alquanto deficitaria ed obsoleta.
Fino a qualche anno fa la ricerca progettuale di soluzioni per l’accessibilità pubblica o privata si è riferita, più o meno consapevolmente, alle esigenze di due tipologie di utenza molto specifiche e caratterizzate da una certa stabilità e omogeneità dei parametri funzionali. Le disabilità motorie (para e tetraplegia) e visive (ipovisione di varia natura e cecità) sono state per anni il riferimento principe delle ricerche, tanto che i testi di legge più importanti vigente in Italia (la Legge n. 13 del 1989 e il conseguente Decreto Ministeriale 286/1989), possono essere considerati come quasi esclusivamente destinati alla loro tutela. Per più di venti anni in Italia si è ritenuto un edificio accessibile se coerente a quanto stabilito da questa normativa, fatto che ha contribuito, al di là delle intenzioni di chi l’ha promulgata, a costruire una cultura dell’accessibilità parziale ed incompleta.
Oggi sappiamo che ogni persona presenta esigenze diverse in funzione delle diverse condizioni in cui si trova, nelle diverse fasi della sua vita. Sappiamo che la disabilità della persona è determinata non solo dalle sue caratteristiche e funzionalità fisiologiche, ma anche, e soprattutto, dal diverso grado di supporto del contesto in cui si trova a vivere.
Nel corso della ricerca hai avuto molti momenti di scambio diretto con le famiglie della comunità Duchenne, anche su aspetti molto pratici legati alla progettazione. Come è stato confrontarsi con loro?
Lo scambio con le famiglie è stato determinante per la buona riuscita del lavoro. I ragazzi DMD con le loro famiglie sono stati i protagonisti assoluti della ricerca. È per loro che la ricerca è stata pensata; è grazie a loro che la ricerca ha preso via del tutto inesplorate e nuove; è grazie a loro che si sono definiti bisogni ed esigenze specifici per cercare di colmare lacune od ostacoli della vita quotidiana.
L’intero lavoro ha come principale obiettivo la loro soddisfazione.
Il viaggio di conoscenza con le famiglie è stato intenso, a volte anche duro, ma sicuramente mi ha dato molta soddisfazione.
Le famiglie, nel momento in cui viene comunicata loro la diagnosi e malattia del figlio, si trovano a dover compiere (o per lo meno conoscere) azioni complesse riguardo al benessere abitativo e psicologico per garantire una vita qualitativa al proprio figlio.
Pertanto consiglio di iniziare a ragionare sull’accessibilità domestica il prima possibile. È vero che la tecnologia e le prassi mediche si evolvono ad una velocità sorprendente, ma aspettare l’ultimo momento (per garantirsi la migliore e più nuova tecnologia presente sul mercato commerciale e per possedere maggiore sicurezza sulla stato degenerativo della malattia), non paga a lungo termine, a mio parere. È bene che un bambino DMD cresca relazionandosi fin da subito con un contesto inclusivo e flessibile. La scelta di effettuare lavori all’ultimo momento ha una ricaduta micidiale sulla psiche del ragazzo, che vede il modificarsi degli ambienti domestici per sua “responsabilità”.
La ricerca ci insegna che non è così.
Il più delle volte è il contesto nel quale viviamo che mette maggiormente in risalto i nostri limiti o i nostri pregi; accorgimenti come i sottotitoli nei programmi televisivi hanno reso, ad esempio, inclusiva una società dotata di tale tecnologia per le persone affette da sordità.
Come vedi il tuo futuro? Vorresti continuare a lavorare nell'ambito della progettazione per l'accessibilità, in particolare in relazione all'ambito della DMD?
Sicuramente cercherò di continuare a progettare e pensare per le persone e per il loro benessere.
La società, anche a causa dell’invecchiamento della popolazione, si sta sensibilizzando sui temi riguardanti l’accessibilità fisica, cognitiva e sociale e si stanno svolgendo ricerche interessanti per garantire autonomia alle persone con disabilità temporanee o permanenti.
Grazie all’attualità del tema analizzato, le possibili aree di ricerca future sono molto vaste ed eterogenee. I manuali ad oggi elaborati sono limitati alla descrizione e prescrizione degli spazi interni all’abitazione; la necessità e l’intenzione sarebbe quella di proseguire il lavoro, sviluppando e completando quello già svolto, su tematiche inerenti gli accessi, i trasferimenti orizzontali e verticali, le pertinenze, il garage, gli spazi comuni in condomini o palazzi. Tali tematiche sono di vitale importanza per integrare il prodotto attuale e creare uno strumento completo per famiglie e tecnici professionisti, che sia di ausilio nella corretta definizione degli spazi interni ed esterni della casa.
Un’ulteriore area di indagine potrebbe essere rivolta ad ampliare le indicazioni fornite agli spazi pubblici e commerciali. Il tema dell’accessibilità e dell’inclusione sociale è in rapido sviluppo e dovrebbe rappresentare un punto di riferimento per le future scelte politiche ed istituzionali. A causa del cambiamento demografico degli ultimi anni, in Europa l’età media dei cittadini si sta sempre più alzando e perciò riuscire a garantire spazi e servizi pubblici e privati accessibili ed inclusivi dovrà essere una priorità a beneficio di tutte le persone, disabili o normodotate. In uno scenario di questo tipo, il design potrebbe e dovrebbe avere un ruolo importante in quanto riuscire a progettare in funzione dei bisogni, necessità ed aspettative delle persone è garanzia di qualità; se a questi aspetti sociali e culturali affianchiamo le innovazioni del settore ICT, il raggiungimento dei risultati è garantito.
Per concludere ci tengo a ringraziare pubblicamente Parent Project onlus per l’opportunità datami, sperando di continuare a collaborare per cercare di migliorare la qualità della vita delle famiglie Duchenne.
Catabasis Pharmaceuticals presenta i risultati positivi su CAT-1004 provenienti dalla parte A dello studio clinico MoveDMD alla conferenza clinica 2016 della Muscular Dystrophy Association.
- Lo studio ha dimostrato sicurezza, tollerabilità e farmacocinetica favorevole nei pazienti
- I risultati supportano l'inizio della parte B dello studio previsto per la prima metà del 2016
21 marzo 2016
CAMBRIDGE, Mass --(BUSINESS WIRE)--Catabasis Pharmaceutical Inc una company biofarmaceutica allo stadio clinico, ha annunciato oggi i dati positivi provenienti dalla parte A del trial MoveDMD con CAT-1004 per la Distrofia Muscolare di Duchenne (DMD). L'obiettivo della parte A era stabilire la sicurezza, tollerabilità e farmacocinetica nei ragazzi DMD tra i 4 e i 7 anni per supportare l'avvio della parte B dello studio MoveDMD, un trial di 12 settimane finalizzato a valutare l'efficacia. Tutti e tre i dosaggi di CAT-1004 testati sono stati generalmente ben tollerati senza segnali di sicurezza osservati. La gran parte degli effetti avversi sono stati di natura lieve e gli effetti avversi osservati sono stati diarrea (4 eventi) feci morbide (3 eventi) e dolore nella parte alta dell'addome (2eventi). Non ci sono stati eventi avversi gravi e nessuna interruzione dell'assunzione della molecola. Sei dei 9 eventi avversi totali sono stati osservati nel gruppo al più alto dosaggio. Non ci sonno state tendenze o problemi di sicurezza negli esami fisici o di laboratorio, EKG o segni vitali.
Dai dati farmacocinetici concludiamo che i dosaggi di 33mg/kg somministrati 2 o 3 volte al giorno sono attesi raggiungere l'esposizione sistemica alla quale è stata osservata l'inibizione di NF-kB negli adulti. La farmacocinetica ha dimostrato un aumento dell'esposizione dose-dipendente e un effetto modesto nella composizione dei pasti. Con dosi singole di 33mg/kg c’erano differenze minime sia nell'area sotto la curva (AUC) che nella massima concentrazione nel siero (Cmax) quando CAT-1004 è stato somministrato con un pasto sia ad alto contenuto di grassi che a basso contenuto. In base a questi risultati, le dosi giornaliera selezionate per la parte B saranno 67 o 100 mg/kg somministrata con il cibo a 33mg/kg 2 o 3 volte al giorno. Da questi risultati Catabasis pianifica di iniziare la parte B del trial MoveDMD nella prima metà del 2016, dipendentemente dalle approvazioni regolatorie del protocollo proposto. Il Parent Project Muscular Dystrophy e la Muscular Dystrophy Association stanno fornendo i finanziamenti per supportare i viaggi dei partecipanti per il trial MoveDMD.
Joanne Donovan, M.D., Ph.D., Chief Medical Officer di Catabasis ha commentato “I risultati favorevoli relativi alla sicurezza, tollerabilità e farmacocinetica nei ragazzi affetti da DMD sono incoraggianti e siamo impazienti di iniziare la parte B del trial MoveDMD. CAT-1004 è una potenziale terapia orale per la DMD in grado di modificare la patologia indipendentemente dal tipo di mutazione nella ditrofina. Abbiamo ricevuto un sostegno significativo e entusiasmo per il nostro trial MoveDMD e CAT-1004 dalle famiglie, le associazioni di pazienti, i ricercatori e lo staff dello studio e li ringraziamo per la loro partecipazione e sostegno”.
Richard Finkel, M.D., Division Chief, Division of Neurology, Department of Pediatrics presso il Nemours Children’s Health System ha commentato “Il bisogno medico insoddisfatto nella DMD è profondo e potenziali terapie che potrebbero fare una differenza significativa sono necessarie. Aver mostrato risultati di sicurezza, tollerabilità e farmacocinetica positivi è una tappa importante nello sviluppo di CAT-1004. Sono impaziente degli avanzamenti di questa nuova potenziale terapia.
CAT-1004 è una piccola molecola per uso orale che la company crede abbia la potenzialità di essere una terapia in grado di modificare la patologia per il trattamento della DMD, indipendentemente dalla mutazione nella distrofina che ha causato la patologia. CAT-1004 è un inibitore di NF-kB, una proteina che è cronicamente attivata nella DMD così come in molte altre patologie muscolo scheletriche e in patologie rare. Nei modelli animali della DMD CAT-1004 ha inibito NF-kB riducendo la degenerazione muscolare e aumentando la rigenerazione.
Lo studio MoveDMD viene condotto in due parti sequenziali. La parte A e quella B. Nella parte A del trial MoveDMD, 17 bambini deambulanti tra i 4 e i 7 anni con una diagnosi geneticamente confermata di DMD e con una gamma di mutazioni nel gene della distrofina, hanno ricevuto CAT-1004. I ragazzi non avevano mai assunto steroidi o non li avevano presi per almeno sei mesi prima dello studio. La parte A del trial si è svolta in tre siti negli Stati Uniti e ha valutato la sicurezza tollerabilità e farmacocinetica di CAT-1004 nei pazienti a tre livelli di dosaggio (33mg/kg/giorno, 67 mg/kg/giorno e 100 mg/kg/giorno) nel corso di 7 giorni di somministrazione. La parte B sarà uno studio randomizzato, in doppio cieco e controllato con placebo per valutare la sicurezza e l'efficacia di CAT-1004 nella DMD nel corso di un periodo di 12 settimane presso 5 centri clinici dello studio negli Usa a due livelli di dosaggio, 67 mg/kg/giorno e 100mg/kg/giorno, soggetto ad approvazione regolatoria. Ai ragazzi che hanno partecipato alla parte A dello studio MoveDMD sarà chiesto di partecipare alla parte B e ulteriori partecipanti saranno anche arruolati fino a circa 30 ragazzi. Attualmente stiamo identificando gli ulteriori pazienti che sono interessati a partecipare alla parte B del trial. I criteri d'inclusione si prevede siano simili a quelli della parte A.
Ulteriori informazioni sul trial MoveDMD sono disponibili alla pagina "clinical trials" del sito web di Catabasis e su ClinicalTrial.gov al numero di identificazione dello studio NCT02439216.
Traduzione a cura dell’Ufficio Scientifico di Parent Project onlus
