


Progetto teleriabilitazione: il calendario di maggio
Prosegue il progetto dedicato alla fisioterapia online, realizzato grazie al supporto di Cleverage e PTC Therapeutics. Tramite il progetto è possibile usufruire di consulti fisioterapici a distanza a supporto dei programmi riabilitativi prescritti, intesi come assistenza alla famiglia o al caregiver durante lo svolgimento degli esercizi domiciliari oppure come momento di confronto specifico per ogni famiglia, a seconda delle proprie esigenze.
E’ disponibile il calendario delle sedute di teleriabilitazione relativo al mese di maggio (fasce orarie h 17-18 e h 18-19):
- DMD 0-5 anni: Martedì 3/05 - Venerdì 6/05
- DMD 5-10 anni: Lunedì 9/05 - Martedì 10/05 - Venerdì 13/05
- DMD 10-15 anni: Lunedì 16/05 - Martedì 17/05 - Venerdì 20/05
- DMD >15 anni: Lunedì 23/05 - Martedì 24/05 - Venerdì 27/05
Per partecipare ai teleconsulti sarà necessario iscriversi contattando Cristina Bella, fisioterapista Parent Project, via mail all’indirizzo c.bella@parentproject.it oppure tramite Whatsapp al numero 340 154 0979, specificando nome, cognome, età di vostro figlio, indirizzo mail per l’iscrizione su Zoom e numero di telefono su cui essere contattati. Vi verranno forniti maggiori dettagli sullo svolgimento del progetto e il calendario specifico per la vostra fascia d’età per poter scegliere la data più comoda a voi tra quelle disponibili.
Scarica il dépliant del progetto:

Inibizione del fattore di trascrizione Nfix farmaco-mediata come nuova terapia per la distrofia muscolare di Duchenne - Report Finale
Finanziato da Parent Project aps, il progetto coordinato da Graziella Messina è uno dei quattro che ha ricevuto, attraverso il bando per la ricerca 2018, la tipologia di finanziamento detta Fast Track per l’importo di 20.000€. Il progetto è iniziato a febbraio del 2019 e ha avuto una durata di due anni. L'idea innovativa a cui si è dedicato il team di ricerca è stata quella di rallentare la rigenerazione muscolare favorendo un accumulo di fibre muscolari con un metabolismo ossidativo, note per essere più resistenti ai danni associati alla patologia. Il gruppo di ricerca della Professoressa Messina ha dimostrato che l’assenza del fattore di trascrizione Nfix, Nuclear Factor One, rallenta la rigenerazione e converte tutte le fibre muscolari a fibre a contrazione lenta.
I processi molecolari alla base dell’insorgenza e progressione delle distrofie muscolari non sono ancora completamente noti. Sebbene se ne conoscano le basi genetiche, la distrofia muscolare di Duchenne è una patologia complessa, nella quale diversi aspetti (come infiammazione, rigenerazione/degenerazione muscolare, tipologia dei muscoli interessati) compartecipano nel determinare il quadro diagnostico e di progressione. Comprendere questa complessità e i fattori coinvolti nella patologia consentirà di aumentare l’efficienza delle future terapie cellulari e geniche in corso di sviluppo.
Il fattore di trascrizione Nfix (Nuclear Factor One) è una delle proteine responsabili della progressione della distrofia muscolare di Duchenne. Nel laboratorio della Professoressa Messina è stato dimostrato che l’assenza di Nfix a livello genetico in topi distrofici determina un miglioramento sia fisiologico sia funzionale dei muscoli: minore infiammazione, minore degenerazione muscolare, minore fibrosi, migliore prestazione fisica.
L’obiettivo del gruppo di ricerca è stato quello di sviluppare un approccio farmacologico per inibire la proteina Nfix e ottenere un miglioramento istologico e funzionale nella distrofia muscolare di Duchenne.
Per inibire farmacologicamente una proteina è necessario conoscere bene quali sono i circuiti molecolari che la regolano nelle cellule, per poi potere interferire con essi dall’esterno.
Qualche anno fa, il gruppo di ricerca ha scoperto una delle vie di segnalazione che regolano i livelli del fattore Nfix nelle cellule muscolari: durante lo sviluppo e le fasi post-natali dei mioblasti, Nfix è regolato dalle MAP chinasi, in particolare dall’enzima ERK.

Grazie al contributo di Parent Project aps, il team ha deciso di indagare circa il possibile riutilizzo nella Duchenne del farmaco Trametinib (GSK) – già utilizzato in clinica per il trattamento del melanoma metastatico – in grado di inibire le MAP chinasi e ridurre l’attività dell’enzima ERK. Somministrando questo farmaco prima in culture cellulari in vitro e poi in un modello distrofico, il Sarcoglicano null, è stato osservato un buon livello di inibizione di Nfix. In particolare, topi distrofici adulti (topi Sgca null) trattati con il Trametinib per via orale tutti i giorni per 14 giorni a due dosi diverse (3 e 6 mg/kg) hanno mostrato una buona riduzione di Nfix (circa il 20%) nei muscoli rispetto al controllo.
Inoltre, i muscoli trattati con Trametinib presentavano un maggior numero di fibre muscolari lente a metabolismo ossidativo, note per essere più resistenti ai danni associati alla patologia. Tuttavia, per gli altri parametri morfologici muscolari analizzati (calibro delle miofibre, livelli di rigenerazione e necrosi, estensione aree di infiammazione, fibrosi) i ricercatori non hanno riscontrato dei cambiamenti significativi rispetto al gruppo controllo, ai dosaggi e tempistiche seguiti.
In modo completamente inatteso hanno, purtroppo, notato l’aumento di calcificazioni muscolari in seguito al trattamento con il farmaco. Hanno riscontrato tali alterazioni istologiche soltanto nei topi distrofici trattati e non in topi sani (wild-type) a cui sono stati somministrati gli stessi dosaggi di Trametinib alle stesse tempistiche.
Le calcificazioni muscolari indotte dal trattamento cronico del Trametinib non erano state mai descritte finora per il farmaco, i cui trial pre-clinici e clinici precedenti (necessari per il suo utilizzo in clinica) sono stati condotti su modelli animali e volontari/pazienti non affetti da distrofia muscolare di Duchenne.
Questo aspetto ha chiaramente rappresentato un ostacolo all’ulteriore sviluppo dello studio, limitando la possibilità di aumentare il dosaggio e/o le tempistiche di somministrazione del Trametinib che è in grado di diminuire Nfix nei muscoli distrofici in maniera non sufficiente a ottenere il miglioramento istologico sperato.
I risultati di questo progetto, sebbene non completamente positivi, hanno permesso di fare luce sulla regolazione di Nfix da parte delle MAPK ed ERK in un contesto distrofico.
Ciò consentirà di approfondire il legame tra ERK ed Nfix, riuscendo a trovare una interazione più diretta e testare farmaci sempre più specifici. Inoltre, in un processo di pura serendipity, tale correlazione tra ERK, distrofia e calcificazioni muscolari (mai descritta finora) apre le porte a numerose linee di ricerca per la comprensione dei meccanismi molecolari e cellulari alterati nella distrofia muscolare.

Riprogrammazione metabolica: una nuova strategia terapeutica per la distrofia muscolare di Duchenne - Report Finale
Finanziato da Parent Project aps, il progetto coordinato da Silvia Consalvi è uno dei tre che ha ricevuto, attraverso il bando per la ricerca 2018, la tipologia di finanziamento detta Grant Application, per l’importo di 80.000€. Il progetto è iniziato ad aprile del 2019 e ha avuto una durata di due anni. Il team di ricerca ha testato per la prima volta l’efficacia di un farmaco – SRT2104 – che potrebbe contribuire a estendere nel tempo l’efficacia di givinostat includendo, quindi, pazienti con distrofia muscolare di Duchenne in fase avanzata.
Il trattamento farmacologico della distrofia muscolare di Duchenne (DMD) con inibitori delle istone deacetilasi (HDACi) è attualmente in fase di sperimentazione in studi clinici di fase 3 con il farmaco givinostat; tuttavia, studi preclinici in modelli animali della DMD (topi mdx) hanno indicato che gli effetti benefici degli HDACi sono limitati agli stadi iniziali della malattia e vengono mediati prevalentemente da una popolazione cellulare, i progenitori fibroadipogenici (FAPs), che si trova negli interstizi del tessuto muscolare, dove svolge un ruolo chiave durante la rigenerazione muscolare (Mozzetta et al., 2013; Saccone et al., 2014). In particolare, gli HDACi inducono i FAPs a mantenere un’azione pro-rigenerativa e prevengono la loro conversione in effettori cellulari della degenerazione fibro-adiposa dei muscoli. Tuttavia questi effetti terapeutici degli HDACi sono circoscritti alle fasi precoci della patologia. La perdita degli effetti benefici degli HDACi, osservata negli stadi più avanzati della DMD, è almeno in parte dovuta allo sviluppo di una resistenza dei FAPs agli HDACi (Mozzetta et al., 2013; Saccone et al., 2014).
Il primo obiettivo di questo progetto è stato, pertanto, spiegare i meccanismi biologici alla base di tale resistenza, al fine di sviluppare nuove strategie terapeutiche che possano allungare la finestra temporale di azione degli HDACi.
Gli studi del team della dottoressa Consalvi hanno dimostrato che i FAPs di topi mdx in fase avanzata della patologia esibiscono un'attività HDAC aberrante e, conseguentemente, alterazioni a livello del genoma dell'acetilazione istonica che conferiscono una parziale resistenza al trattamento con HDACi (Consalvi et al., 2021). I FAPs mdx a stadio avanzato mostrano, infatti, una minore acetilazione sui promotori di geni coinvolti nella proliferazione, associata a una ridotta espressione genica. Queste alterazioni non possono essere contrastate dal trattamento con HDACi, a causa di una resistenza generale all'iperacetilazione indotta dagli HDACi. Al contempo, FAPs mdx a stadio avanzato esibiscono una maggiore acetilazione ed espressione dei geni coinvolti nel fenotipo secretorio associato alla senescenza (SASP). Sorprendentemente, su questa categoria di geni gli HDACi riducono i livelli di acetilazione ed espressione genica.
Questi dati rivelano che, durante la progressione della DMD, i FAPs sviluppano caratteristiche associate alla patologia che ricordano la senescenza, ovvero l’invecchiamento cellulare. La ridotta proliferazione e l’attivazione della SASP nei FAPs risultano, inoltre, eventi distinti correlati alla patologia e farmacologicamente dissociabili. Infatti, mentre gli HDACi falliscono nel ripristinare le capacità proliferative dei FAPs, riescono a inibire la SASP suggerendo che il trattamento possa mantenere l’effetto anti-fibrotico e anti-infiammatorio anche a stadi tardivi della patologia (Consalvi et al., 2021).
La proliferazione cellulare è un meccanismo biologico dal grande dispendio energetico e dunque intimamente legato alle capacità metaboliche delle cellule. Studi precedenti hanno analizzato la connessione tra proliferazione e metabolismo nei FAPs (Reggio et al., 2020). I dati hanno rivelato che durante la progressione della DMD, i FAPs sviluppano alterazioni nella funzionalità dei mitocondri, organelli che rappresentano la principale fonte di energia delle cellule. Tale deficit funzionale determina un aumento dello stress ossidativo che rafforza la SASP e compromette le capacità proliferative dei FAPs. In questo studio è stato, quindi, valutato il trattamento farmacologico con SRT2104, attivatore della Sirtuina1, quale agente in grado di promuovere l’attività mitocondriale. I risultati delle analisi hanno dimostrato che SRT2104 è stato in grado di incrementare le capacità metaboliche dei FAPs a stadi tardivi della DMD riportandole ai livelli dei FAPs a stadi precoci. Inoltre, il trattamento combinato di SRT2104 con givinostat ha permesso di recuperare l’attività mitocondriale e ridurre la SASP, ripristinando il tasso proliferativo dei FAPs. Questi dati suggeriscono che i due trattamenti farmacologici possano sinergizzare, determinando un “ringiovanimento” delle FAPs in grado di estendere gli effetti pro-rigenerativi degli HDACi anche a pazienti in stadi avanzati della DMD.

Rappresentazione schematica dell’effetto di HDACi e SRT2104 sui FAPs durante la progressione della DMD.
Referenze
Mozzetta C, Consalvi S, Saccone V, Tierney M, Diamantini A, Mitchel KJ, Marazzi G, Borsellino G, Battistini L, Sassoon D et al (2013). Fibroadipogenic progenitors mediate the ability of HDAC inhibitors to promote regeneration in dystrophic muscles of young, but not old mdx mice. EMBO Mol Med 5: 626-639
Saccone V, Consalvi S, Giordani L, Mozzetta C, Barozzi I, Sandoná M, Ryan T, Rojas-Muñoz A, Madaro L, Fasanaro P et al (2014) HDAC-regulated myomiRs control BAF60 variant exchange and direct the functional phenotype of fibro-adipogenic progenitors in dystrophic muscles. Genes Dev 28: 841-857
Consalvi S, Tucciarone L, Macrì E, De Bardi M, Picozza M, Salvatori I, Renzini A, Valente S, Mai A, Moresi V, Puri PL (2021). Partial resistance to HDAC inhibitors in FAPs of dystrophic muscles at late stages of disease is associated to epigenetic and transcriptional features of cellular senescence. BioRxiv doi: https://doi.org/10.1101/2021.04.26.441412
Reggio A, Rosina M, Krahmer N, Palma A, Petrilli LL, Maiolatesi G, Massacci G, Salvatori I, Valle C, Testa S et al (2020) Metabolic reprogramming of fibro/adipogenic progenitors facilitates muscle regeneration. Life Sci Alliance 3(3):e202000646
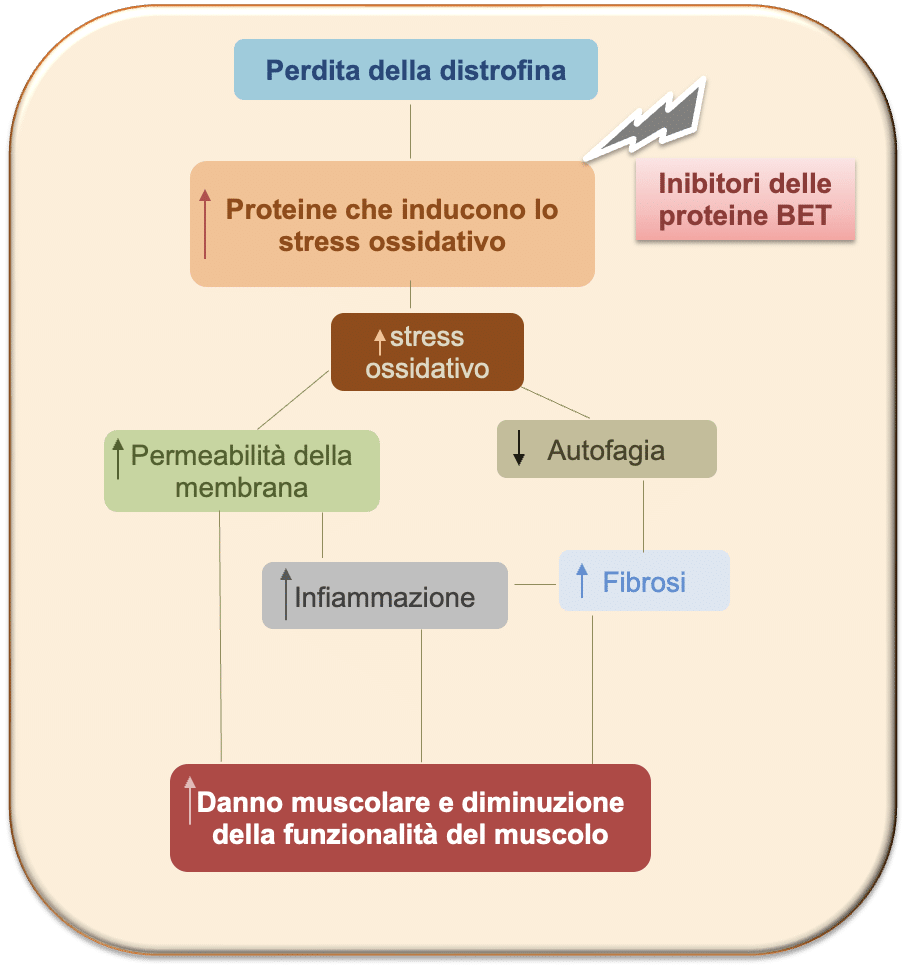
UN NUOVO APPROCCIO EPIGENETICO PER MIGLIORARE L’OMEOSTASI NELLA DISTROFIA MUSCOLARE DI DUCHENNE - REPORT FINALE
Finanziato da Parent Project aps, il progetto coordinato da Giuseppina Caretti è uno dei quattro che ha ricevuto, attraverso il bando per la ricerca 2018, la tipologia di finanziamento detta Fast Track, per l’ importo di 20.000€. Il progetto è iniziato a luglio del 2019 e ha avuto una durata di due anni. Precedenti studi del gruppo di ricerca hanno dimostrato che la somministrazione degli inibitori BET migliora la funzionalità e lo stato del muscolo in un modello per la distrofia muscolare di Duchenne (il topo mdx). L’obiettivo è stato capire se gli inibitori BET riducono la fibrosi nel modello di topo mdx e se il trattamento vada a influenzare l’azione delle cellule pre-fibrotiche. Gli inibitori BET sono una classe di farmaci che bloccano una famiglia di proteine contenenti bromodomini e domini extra-terminali (BET).
Nel secondo anno del progetto finanziato da Parent Project aps, il team della professoressa Caretti ha focalizzato l’attenzione su due modelli di studio:
- il modello murino mdx a un’età di 12 mesi, per verificare se gli effetti migliorativi del trattamento con l’inibitore di JQ1 (molecola in grado di prevenire l’atrofia muscolare e di ridurre i livelli di infiammazione) fossero osservabili anche quando alcune caratteristiche fisiopatologiche della patologia, quali la fibrosi, sono più evidenti
- un modello cellulare, derivato da cellule muscolari immortalizzate derivate da biopsie di pazienti affetti dalla distrofia muscolare di Duchenne.
Studiando l’impatto degli inibitori delle proteine BET in topi mdx di 12 mesi, in cui l’accumulo fibrotico è particolarmente accentuato, sono state osservate una riduzione dei livelli di proteine coinvolte nella fibrosi e la diminuzione della deposizione di fibre di collagene nei muscoli tibiali. Le analisi morfologiche con colorazioni particolari e analisi dei trascritti hanno confermato una riduzione della fibrosi dopo la somministrazione dell’inibitore. In questa fase della progressione della patologia, il trattamento con gli inibitori BET ha anche portato a una riduzione dei livelli di marcatori infiammatori quali TNFα e IL6, confermando i dati ottenuti in topi mdx più giovani e i risultati di altri gruppi di ricerca, che hanno mostrato come le proteine BET abbiano un ruolo fondamentale nel promuovere la risposta infiammatoria, in diverse patologie e contesti cellulari.
Per studiare i meccanismi molecolari alla base della riduzione dei livelli di infiammazione e fibrosi, sono state valutate le modulazioni di alcuni fattori trascrizionali ed epigenetici, coinvolti nella regolazione di molecole coinvolte in questi processi;in particolare, le modulazioni del fattore trascrizionale NFkB e della deacetilasi Sirt1, in seguito a trattamento con gli inibitori delle proteine BET, nel modello mdx e nei modelli cellulari.
Infine, è stato utilizzato il modello derivato da cellule immortalizzate di mioblasti di pazienti Duchenne, al fine di confermare che i meccanismi di regolazione mediata dalle proteine BET, precedentemente osservate nel modello mdx, fossero rilevabili anche in questo modello cellulare. In particolare, l’attenzione del team di ricerca si è soffermata sul ruolo delle proteine BET nella regolazione dei livelli dei regolatori dello stress ossidativo e delle molecole pro-infiammatorie.
L’aumento dello stress ossidativo è un evento iniziale nello sviluppo della patologia del muscolo dei pazienti Duchenne e, a sua volta, causa necrosi delle cellule muscolari, inducendo danno muscolare e promuovendo l’infiltrato infiammatorio. Nel complesso, i dati di questo studio mostrano come quest’aumento dello stress ossidativo sia mediato, almeno in parte, dalle proteine BET, in particolare dalla proteina BRD4, che regola in modo diretto i livelli di proteine che portano alla formazione dei radicali liberi. Come noto dalla letteratura, l’aumento dello stress ossidativo è anche associato a un blocco del meccanismo di smaltimento di proteine e macromolecole non funzionanti, ossia l’autofagia, elemento caratteristico del muscolo distrofico. In seguito all’aumento dello stress ossidativo e al danno muscolare, incrementa anche l’infiammazione e di conseguenza l’induzione della fibrosi, ossia di diversi elementi che nel complesso portano a un peggioramento del danno muscolare, fino a compromettere la funzionalità del muscolo.
Come mostrato in figura, i dati sperimentali ottenuti nel modello mdx e nei modelli cellulari mostrano come gli inibitori delle proteine BET possano essere considerati modulatori importanti dello stress ossidativo e di conseguenza dei molteplici processi coinvolti nella fisiopatologia della distrofia di Duchenne.


Contrastare l’infiammazione muscolare nella DMD promuovendo l’attivazione dei macrofagi pro-rigenerativi e inibendo quelli infiammatori tramite farmaci che agiscono sul metabolismo cellulare - Report Finale
Il progetto è iniziato a marzo del 2019 ed è stato realizzato grazie alla collaborazione tra Elisabetta Ferraro, Francesca De Santa e Alessio Torcinaro. Finanziato da Parent Project aps, il progetto è uno dei quattro che ha ricevuto, attraverso il bando per la ricerca 2018, la tipologia di finanziamento alla ricerca detta Fast Track, per l’importo di 20.000€. Dati sempre più numerosi dimostrano che il metabolismo dei macrofagi infiammatori è diverso da quello dei macrofagi pro-rigenerativi e anti-infiammatori. L’obiettivo di questo studio è stato quello di stimolare il metabolismo di tipo mitocondriale tipico dei macrofagi anti-infiammatori al fine di contrastare l’infiammazione del muscolo distrofico e, al contempo, di promuoverne la rigenerazione.
Una corretta rigenerazione muscolare richiede la partecipazione di cellule del sistema immunitario definite macrofagi. Subito dopo un danno muscolare, il muscolo viene colonizzato da numerosi macrofagi pro-infiammatori (tipo M1) che poi vengono sostituiti da macrofagi con effetto anti-infiammatorio e rigenerativo (tipo M2). Viceversa, nella DMD, il danno muscolare è continuo, per cui si ha una presenza costante di macrofagi infiammatori, che risulta deleteria e contribuisce alla riduzione della capacità rigenerativa del muscolo scheletrico.
Le popolazioni macrofagiche di tipo M1 e di tipo M2 utilizzano vie metaboliche diverse per ricavare l’energia necessaria alla loro sopravvivenza. L’idea progettuale è stata quindi quella di stimolare i processi metabolici usati dai macrofagi M2 (metabolismo principalmente mitocondriale) per indurre la maturazione macrofagica verso il tipo M2 e contrastare la maturazione verso il tipo M1 infiammatorio, al fine di ridurre l’infiammazione cronica del muscolo distrofico.
A tal scopo è stato valutato, in vitro, l’effetto anti-infiammatorio di cinque molecole note per agire su vari processi metabolici (riboflavina-5’-fosfato/vitamina B2-fosfato, idebenone, ranolazina, trimetazidina e mildronato), che sono state somministrate a macrofagi murini trattati con uno stimolo pro-infiammatorio, il lipopolisaccaride. È stata valutata l’espressione genica di citochine infiammatorie, quali interleuchina-1beta e interleuchina-6, nonché quella della chemochina CCL2 e di molecole associate prevalentemente al fenotipo rigenerativo, come Arginasi-1 e CD206.
Tra le molecole che hanno indotto in vitro una risposta antiinfiammatoria (riboflavina-5’-fosfato, idebenone e ranolazina), la riboflavina-5’-fosfato e l’idebenone sono state testate in vivo utilizzando il modello murino per la distrofia muscolare di Duchenne (il topo mdx) mediante un trattamento a breve e uno a lungo termine. Il trattamento a breve termine è consistito in un mese di somministrazioni giornaliere (a partire da 2 mesi fino ai 3 mesi di età dei topi), mentre quello a lungo termine è consistito nel trattamento breve addizionato di cicli intermittenti (ogni due mesi) di due settimane di somministrazione fino all’età di 9 mesi.
Riboflavina-5’-fosfato (1mg/topo/die; somministrazione intraperitoneale) - In seguito ai trattamenti con riboflavina-5’-fosfato, sia quello breve che quello lungo, il numero di fibre ossidative è aumentato in maniera considerevole (figura 1A) con una riduzione della fibrosi, dell'espressione del collagene e dell'infiltrato infiammatorio. Rispetto al trattamento breve, il trattamento lungo ha mostrato, inoltre, un maggiore e significativo incremento della funzionalità muscolare (figura 1B) e un aumento dei livelli di marcatori anti-infiammatori come Arginasi-1 e CD163. Inoltre, è risultata evidente e statisticamente significativa la riduzione delle fibre necrotiche in seguito al trattamento con riboflavina-5’-fosfato.

Idebenone (1mg/topo/die; somministrazione intraperitoneale) - Nonostante l’idebenone abbia rivelato un potente effetto anti-infiammatorio in vitro su macrofagi umani, il trattamento a breve termine sul modello murino mdx con idebenone non ha mostrato differenze significative dei parametri valutati rispetto agli animali non trattati. Tuttavia, il trattamento a lungo termine con questa molecola ha indotto un incremento del numero di fibre ossidative, anche se in misura minore rispetto alla riboflavina-5’-fosfato, una riduzione della fibrosi e dell'espressione del collagene e, solo nel 50% degli animali, una riduzione delle fibre necrotiche che si traduce in una assenza di riduzione statisticamente significativa delle fibre necrotiche. Infine, anche il trattamento lungo con idebenone, così come quello con riboflavina-5’-fosfato, ha indotto un significativo incremento della funzionalità muscolare.
Rispetto ad altre terapie anti-infiammatorie, l’uso di queste molecole in clinica potrebbe essere vantaggioso per il fatto che esse potrebbero svolgere contemporaneamente due ruoli: i) ridurre l’infiammazione e ii) promuovere la capacità rigenerativa del muscolo. Inoltre, i dati suggeriscono che queste molecole, poiché agiscono sul metabolismo, abbiano un effetto benefico diretto, oltre che sui macrofagi, anche sulle fibre muscolari il cui metabolismo, nei pazienti distrofici, è fortemente alterato.
