
Un trial clinico in vitro: un semplice, economico e attendibile test per predire l’efficacia di una nuova terapia
Titolare del progetto: Giulio Cossu, Università di Manchester (UK)
Durata: 1 anno
Importo: 72.000 euro
Il progetto di ricerca ha lo scopo di valutare il potenziale terapeutico di diverse terapie in un trial in vitro per misurare rapidamente l’efficacia di un terapia prima di arrivare alla sperimentazione sui pazienti. In questi anni sono stati condotti numerosi trial clinici per sviluppare un terapia efficace per la distrofia muscolare di Duchenne. Ogni trial può costare milioni di euro, durare molti anni ed essere estremamente faticoso in termini di stress per pazienti e famiglie. Il gruppo di ricerca del Professor Cossu propone di sviluppare un test in vitro che permetta di verificare contemporaneamente l’efficacia terapeutica di diversi trattamenti. Nel dettaglio, il gruppo cercherà di distinguere cellule normali da cellule distrofiche e di capire se un determinato trattamento può modificare la risposta delle cellule distrofiche rendendole simili a quelle di cellule normali. In altre parole si tratta di un trial miniaturizzato in coltura dove vengono utilizzate cellule di pazienti corrette geneticamente in modi diversi e coltivate insieme alle cellule malate.
E’ noto che aggiungendo in laboratorio delle fibre muscolari a una coltura dei neuroni del midollo spinale di embrioni di topo si può indurre una contrazione muscolare. Da decenni co-colture di miotubi (fibre muscolari multinucleate) e motoneuroni (neuroni localizzati all’interno del sistema nervoso centrale) sono state perciò utilizzate per promuovere la contrazione dei miotubi in laboratorio attraverso segnali elettrici e chimici.
Le cellule distrofiche, non producendo distrofina, non riescono a sopravvivere e muoiono dopo 24 ore mentre quelle normali continuano a contrarsi per 3-4 giorni. Il gruppo di ricerca del Professor Cossu ha validato il sistema coltivando i miotubi distrofici precedentemente corretti con un vettore virale che causa exon skipping dell’esone 51 e hanno osservato che in questo caso le cellule distrofiche continuano a contrarsi come quelle normali, comportandosi come miotubi sani.
Questo saggio, reso disponibile alla comunità scientifica, permetterebbe quindi di valutare, in modo semplice, economico e rapido, se un certo trattamento esercita un effetto significativo sulle cellule distrofiche che possa giustificare l’investimento in una nuova sperimentazione clinica.
Approfondimento
Report finale
UN TRIAL CLINICO SUL CHIP: UN METODO SEMPLICE, ECONOMICO E RIPRODUCIBILE PER PREDIRE L’EFFICACIA DI UNA NUOVA TERAPIA – REPORT FINALE
Il progetto, iniziato a gennaio del 2019, mirava a identificare cambiamenti iniziali dovuti all’assenza di distrofina in cellule muscolari nelle prime fasi del differenziamento in coltura. Questo per poter eventualmente utilizzare questi cambiamenti come indicatori della possibile efficacia di nuovi interventi terapeutici sperimentali, prima di procedere con sperimentazioni su modelli animali e poi eventualmente iniziare un trial clinico. Sono state usate cellule miogeniche umane sane e cellule provenienti da muscoli distrofici (con una mutazione trattabile con lo skipping dell’esone 51). Sono state inoltre utilizzate le stesse cellule distrofiche, dopo correzione genetica con un vettore lentivirale che esprime un piccolo RNA nucleare, U7snRNA, progettato per saltare l’esone 51 del gene della distrofina (Galli et al. In revisione).
La prima osservazione è stata la presenza di flussi di ioni calcio nelle cellule muscolari DMD in coltura, non appena si differenziano, prima che diventino capaci di contrarsi. Le cellule derivate da muscoli sani non mostrano questi flussi. Quando le cellule distrofiche venivano corrette geneticamente così da esprimere la distrofina, i flussi di calcio scomparivano.
Un esempio di questi flussi ionici, visualizzati da un colorante (Fura2) che aumenta di intensità quando lega gli ioni calcio è mostrato in Figura 1.
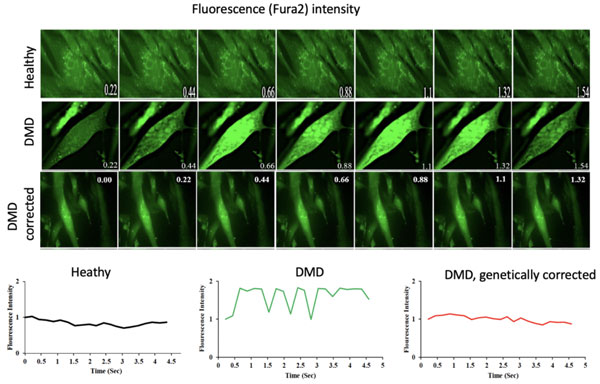
Figura 1. Time frames di filmati di cellule muscolari contenenti Fura2 che mostrano oscillazioni del colore solo nelle cellule distrofiche. L’intensità della fluorescenza è quantificata nel pannello inferiore.
Il passaggio di ioni calcio attraverso le membrane delle fibre muscolari è considerato il meccanismo che innesca una catena di eventi che alla fine porta alla morte delle fibre muscolari e delle cellule del cuore nella distrofia muscolare di Duchenne (DMD) e in altre distrofie muscolari. È considerato un evento tardivo nel meccanismo patogenetico, suscitato dall’allungamento della fibra muscolare durante la contrazione. La novità dell’osservazione del gruppo di ricerca del Professor Cossu consiste nella precocità del flusso di ioni calcio che non può essere causato dalla contrazione perché le cellule muscolari in coltura ancora non si contraggono.
Questi flussi, come atteso, erano bloccati da inibitori dei canali del calcio deputati al rilascio di calcio durante la contrazione muscolare. Nel tentativo di promuovere la maturazione delle cellule muscolari in coltura sono state coltivate insieme a motoneuroni embrionali ed è stato osservato che, in assenza di stimolazione elettrica e di contrazione, i flussi di calcio scomparivano. Il che ha portato a ipotizzare che non l’attività neurale (elettrica) ma il rilascio di qualche molecola da parte dei motoneuroni portasse al blocco dei flussi di calcio. I motoneuroni rilasciano varie molecole quando formano le giunzioni neuro-muscolari, la più studiata delle quali è l’agrina, un proteoglicano (una proteina che lega lunghe catene di zuccheri solforati) che ha un ruolo centrale nella formazione della giunzione ma anche altri ruoli nella maturazione della fibra muscolare. È stata quindi aggiunta soltanto agrina alle colture muscolari ed è stato visto che i flussi di calcio scomparivano. L’agrina forma un legame tra il distroglicano, una glicoproteina cha fa parte del complesso delle proteine associate alla distrofina e quindi è molto ridotta nella DMD e la laminina. Una versione miniaturizzata (mini-agrina) è attualmente considerata come un possibile strumento terapeutico per una forma di distrofia muscolare congenita dovuta all’assenza di una specifica forma di laminina, parte integrante della lamina basale che circonda tutte le fibre muscolari. E’ stato quindi ipotizzato che l’agrina, stabilizzando il legame con la laminina mantenesse il distroglicano in membrana (e forse altre proteine) eliminando così i flussi di calcio.
Il passo successivo è stato capire in che modo. Studi di espressione genica hanno rivelato che la mancanza di distrofina nelle cellule muscolari DMD porta a una ridotta espressione di un sensore di calcio, noto per legarsi e regolare il recettore della rianodina 1. Ciò coincide con la comparsa di picchi di ioni calcio. Le cellule muscolari DMD sane e geneticamente corrette non mostrano questi picchi che sono anche aboliti dagli inibitori del recettore della diidropiridina (DHPR). L’espressione della distrofina mediata da un vettore lentivirale, ma anche l’agrina, stabilizzano il sensore, probabilmente attraverso l’interazione con il distroglicano, e portano alla scomparsa dei flussi transienti di calcio.
Questi dati rivelano un nuovo meccanismo di regolazione del calcio nella DMD e aprono una nuova opportunità terapeutica per bloccare l’afflusso di ioni calcio.